


Therapeutic Effect of Donepezil on Neuroinflammation and Cognitive Impairment after Moderate Traumatic Brain Injury

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. In Vitro Model of TBI
2.2. In Vivo Model of TBI
2.3. In Vitro Study
2.4. In Vivo Study
2.4.1. H&E Staining, Brain Water Content, and Fluoro-Jade B Staining
2.4.2. qRT-PCR and Western Blotting
2.4.3. Cognitive Function Test
2.4.4. Statistical Analysis
3. Results
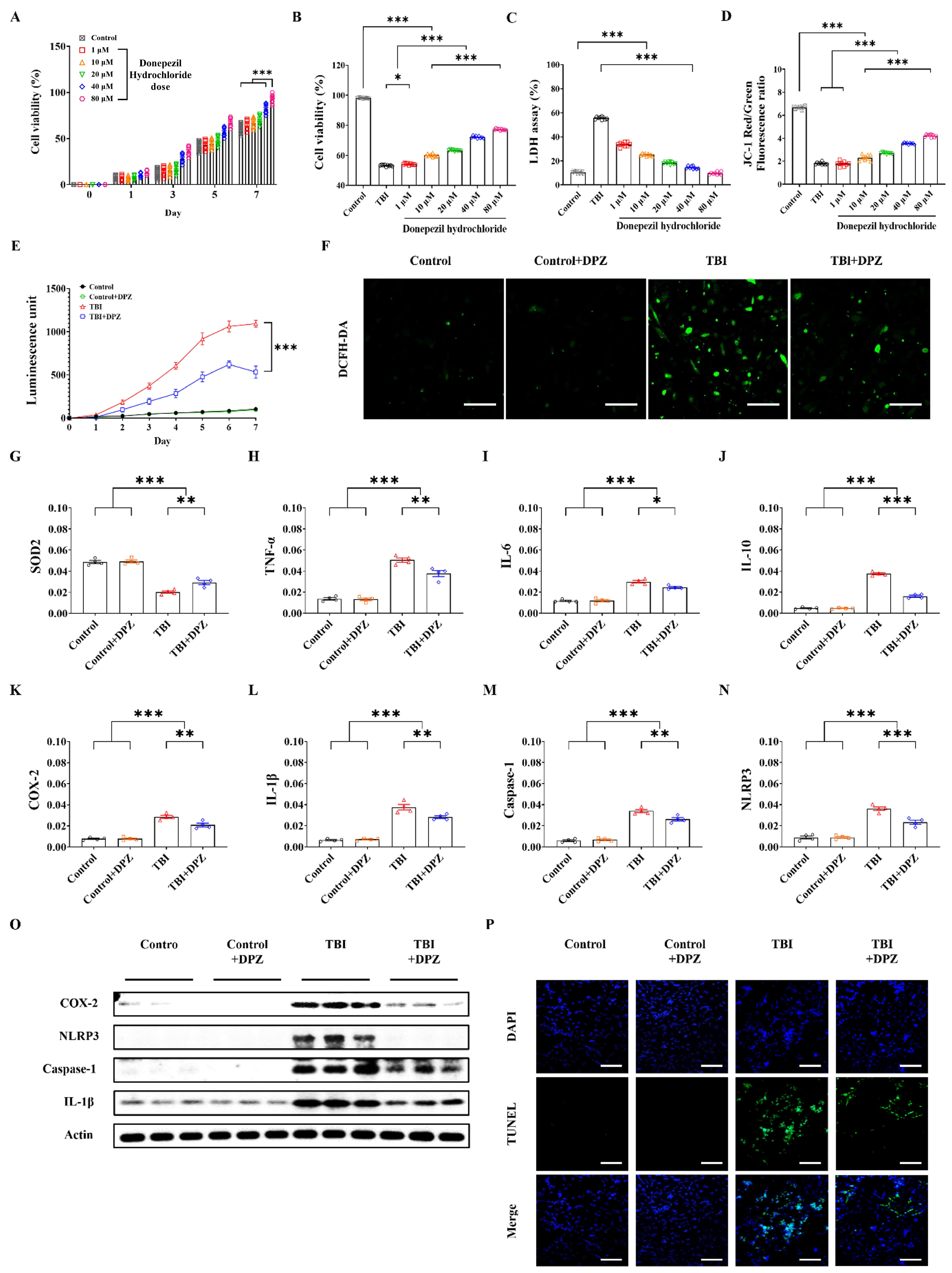
3.1. Cytoprotective Effect in In Vitro TBI
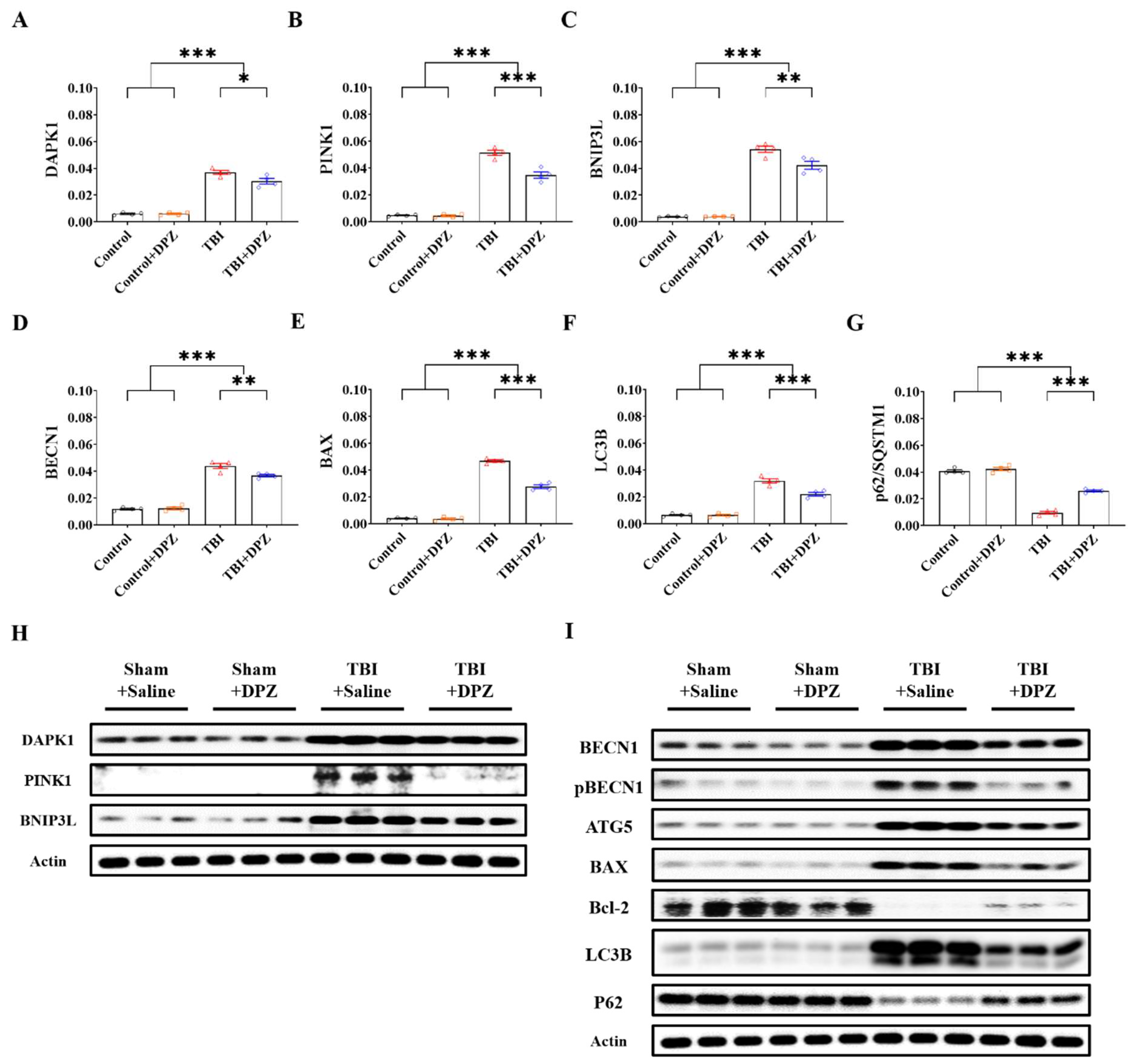
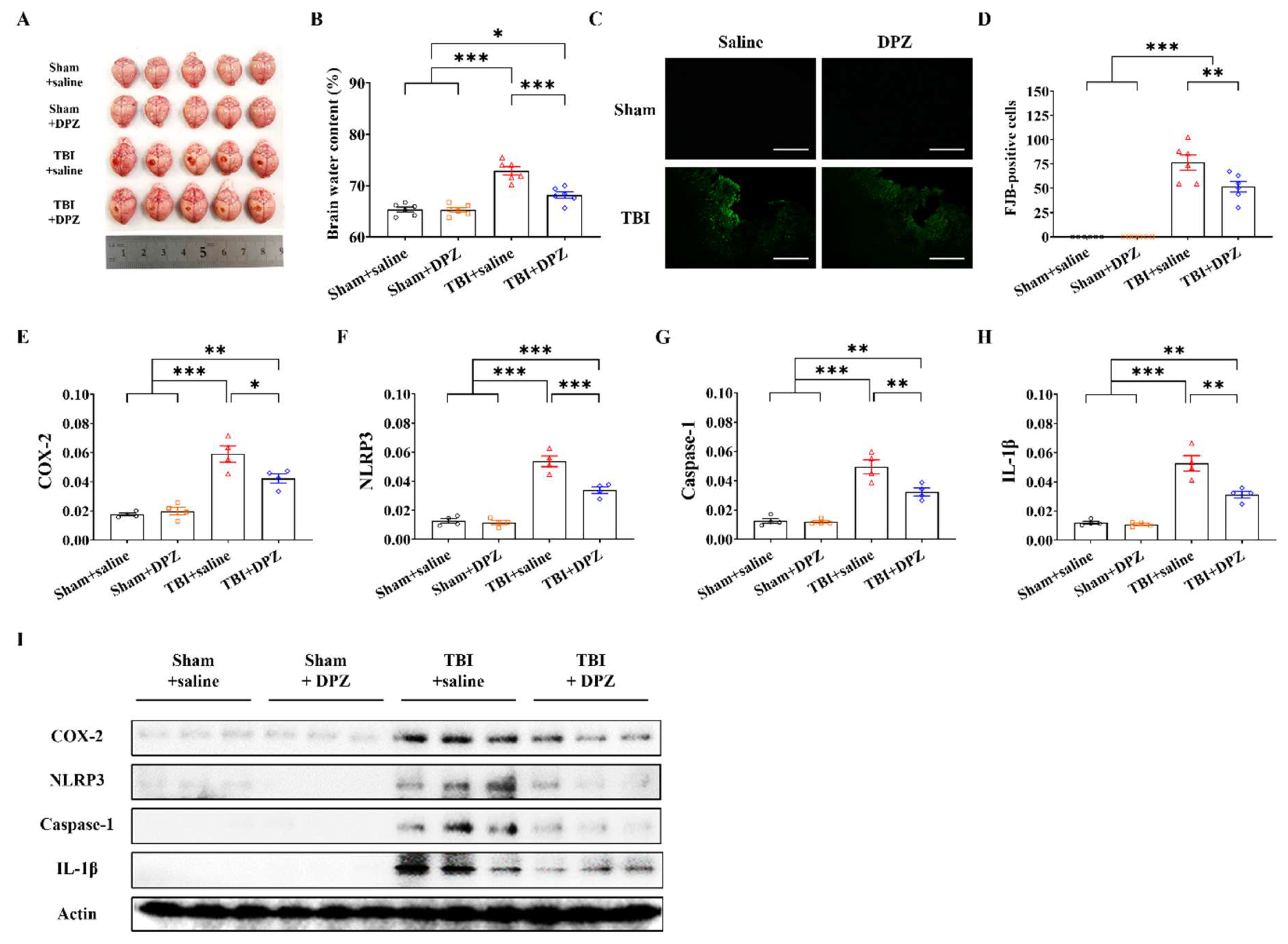
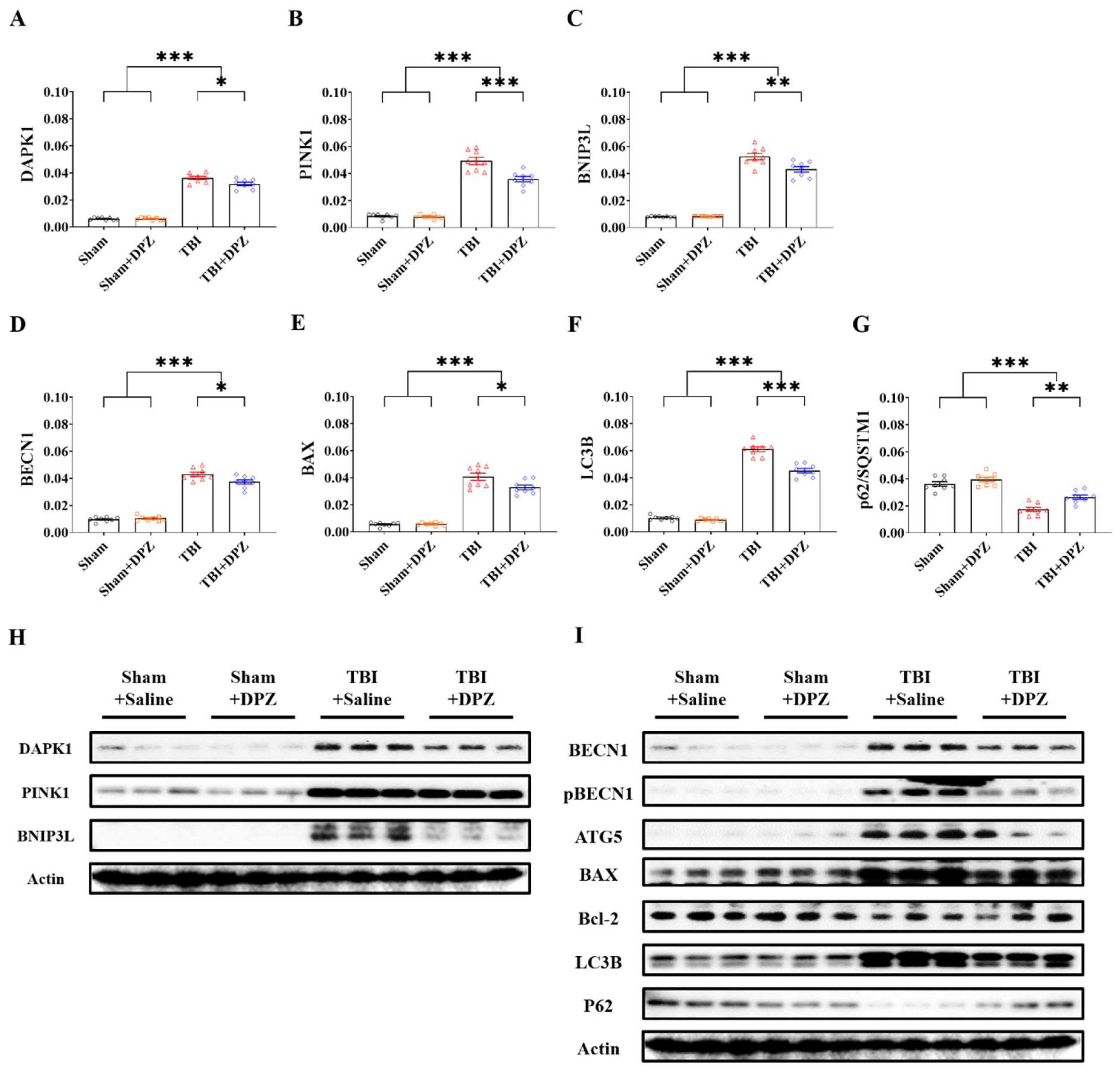
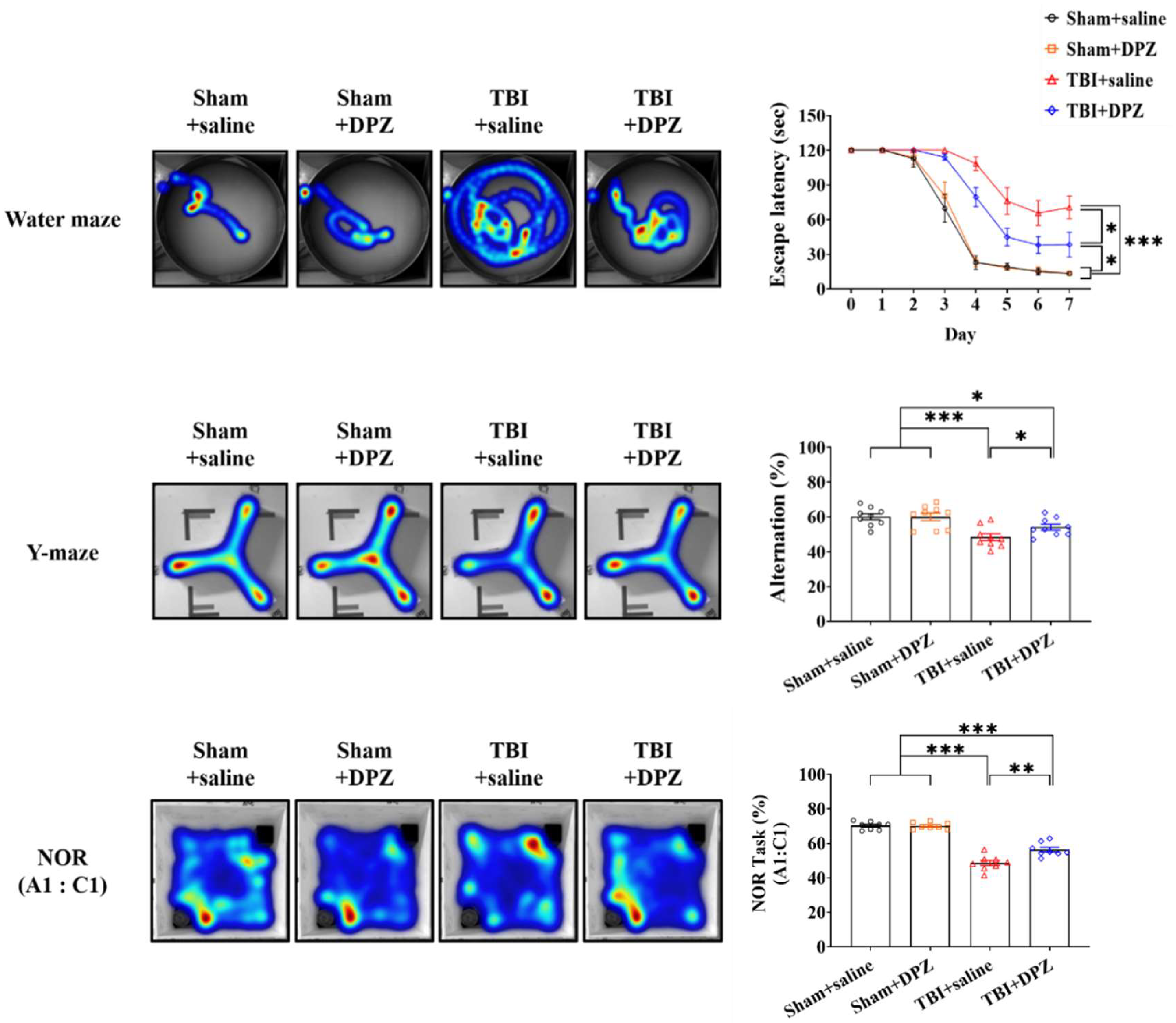
3.2. Neuroprotective Effects in In Vivo TBI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Kim, E.; Park, M.; Jeong, J.; Kim, H.; Lee, S.K.; Lee, E.; Oh, B.H.; Namkoong, K. Cholinesterase Inhibitor Donepezil Increases Mitochondrial Biogenesis through AMP-Activated Protein Kinase in the Hippocampus. Neuropsychobiology 2016, 73, 81–91. [Google Scholar] [CrossRef]
- Ye, C.Y.; Lei, Y.; Tang, X.C.; Zhang, H.Y. Donepezil attenuates Aβ-associated mitochondrial dysfunction and reduces mitochondrial Aβ accumulation in vivo and in vitro. Neuropharmacology 2015, 95, 29–36. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Kojima, A.; Ishikawa, C.; Arai, K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J. Alzheimer‘s Dis. 2010, 22, 295–306. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef]
- Takahashi, K.; Tsuji, M.; Nakagawasai, O.; Katsuyama, S.; Hong, L.; Miyagawa, K.; Kurokawa, K.; Mochida-Saito, A.; Takeda, H.; Tadano, T. Donepezil prevents olfactory dysfunction and α-synuclein aggregation in the olfactory bulb by enhancing autophagy in zinc sulfate-treated mice. Behav. Brain Res. 2023, 438, 114175. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Liu, C.J.; Huang, H.C.; Lin, J.H.; Chen, P.Y.; Su, Y.K.; Chen, C.T.; Chiu, H.Y. A Meta-analysis of Dynamic Prevalence of Cognitive Deficits in the Acute, Subacute, and Chronic Phases After Traumatic Brain Injury. J. Neurosci. Nurs. 2021, 53, 63–68. [Google Scholar] [CrossRef]
- Shin, S.S.; Dixon, C.E. Alterations in Cholinergic Pathways and Therapeutic Strategies Targeting Cholinergic System after Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1429–1440. [Google Scholar] [CrossRef]
- Khateb, A.; Ammann, J.; Annoni, J.M.; Diserens, K. Cognition-enhancing effects of donepezil in traumatic brain injury. Eur. Neurol. 2005, 54, 39–45. [Google Scholar] [CrossRef]
- Morey, C.E.; Cilo, M.; Berry, J.; Cusick, C. The effect of Aricept in persons with persistent memory disorder following traumatic brain injury: A pilot study. Brain Inj. 2003, 17, 809–815. [Google Scholar] [CrossRef]
- Yu, T.S.; Kim, A.; Kernie, S.G. Donepezil rescues spatial learning and memory deficits following traumatic brain injury independent of its effects on neurogenesis. PLoS ONE 2015, 10, e0118793. [Google Scholar] [CrossRef]
- Campbell, K.A.; Kennedy, R.E.; Brunner, R.C.; Hollis, S.D.; Lumsden, R.A.; Novack, T.A. The effect of donepezil on the cognitive ability early in the course of recovery from traumatic brain injury. Brain Inj. 2018, 32, 972–979. [Google Scholar] [CrossRef]
- Walker, W.; Seel, R.; Gibellato, M.; Lew, H.; Cornis-Pop, M.; Jena, T.; Silver, T. The effects of Donepezil on traumatic brain injury acute rehabilitation outcomes. Brain Inj. 2004, 18, 739–750. [Google Scholar] [CrossRef]
- Jeon, J.P.; Kim, S.; Kim, T.Y.; Han, S.W.; Lim, S.H.; Youn, D.H.; Kim, B.J.; Hong, E.P.; Park, C.H.; Kim, J.T.; et al. Association Between Copeptin and Six-Month Neurologic Outcomes in Patients With Moderate Traumatic Brain Injury. Front. Neurol. 2021, 12, 749110. [Google Scholar] [CrossRef]
- Álvarez-Merz, I.; Muñoz, M.D.; Hernández-Guijo, J.M.; Solís, J.M. Identification of Non-excitatory Amino Acids and Transporters Mediating the Irreversible Synaptic Silencing After Hypoxia. Transl. Stroke Res. 2023. [Google Scholar] [CrossRef]
- Wolf, M.S.; Bayır, H.; Kochanek, P.M.; Clark, R.S.B. The role of autophagy in acute brain injury: A state of flux? Neurobiol. Dis. 2019, 122, 9–15. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, L.; Yang, X. Interaction between autophagy and the NLRP3 inflammasome in Alzheimer‘s and Parkinson‘s disease. Front. Aging Neurosci. 2022, 14, 1018848. [Google Scholar] [CrossRef]
- Minchev, D.; Kazakova, M.; Sarafian, V. Neuroinflammation and Autophagy in Parkinson‘s Disease-Novel Perspectives. Int. J. Mol. Sci. 2022, 23, 14997. [Google Scholar] [CrossRef]
- Tan, S.W.; Zhao, Y.; Li, P.; Ning, Y.L.; Huang, Z.Z.; Yang, N.; Liu, D.; Zhou, Y.G. HMGB1 mediates cognitive impairment caused by the NLRP3 inflammasome in the late stage of traumatic brain injury. J. Neuroinflamm. 2021, 18, 241. [Google Scholar] [CrossRef]
- Salvador, E.; Burek, M.; Förster, C.Y. An In Vitro Model of Traumatic Brain Injury. Methods Mol. Biol. 2018, 1717, 219–227. [Google Scholar]
- Youn, D.H.; Han, S.W.; Kim, J.T.; Choi, H.; Lee, A.; Kim, N.; Jung, H.; Hong, E.P.; Park, C.H.; Lee, Y.; et al. Oxiracetam alleviates anti-inflammatory activity and ameliorates cognitive impairment in the early phase of traumatic brain injury. Acta Neurochir. 2023, 165, 2201–2210. [Google Scholar] [CrossRef]
- Youn, D.H.; Tran, N.M.; Kim, B.J.; Kim, Y.; Jeon, J.P.; Yoo, H. Shape effect of cerium oxide nanoparticles on mild traumatic brain injury. Sci. Rep. 2021, 11, 15571. [Google Scholar] [CrossRef]
- Youn, D.H.; Jung, H.; Tran, N.M.; Jeon, J.P.; Yoo, H. The Therapeutic Role of Nanoparticle Shape in Traumatic Brain Injury: An in vitro Comparative Study. J. Korean Neurosurg. Soc. 2022, 65, 196–203. [Google Scholar] [CrossRef]
- Fu, C.; Zhang, X.; Zeng, Z.; Tian, Y.; Jin, X.; Wang, F.; Xu, Z.; Chen, B.; Zheng, H.; Liu, X. Neuroprotective Effects of Qingnao Dripping Pills Against Cerebral Ischemia via Inhibiting NLRP3 Inflammasome Signaling Pathway: In Vivo and In Vitro. Front. Pharmacol. 2020, 11, 65. [Google Scholar] [CrossRef]
- Youn, D.H.; Kim, Y.; Kim, B.J.; Jeong, M.S.; Lee, J.; Rhim, J.K.; Kim, H.C.; Jeon, J.P. Mitochondrial dysfunction associated with autophagy and mitophagy in cerebrospinal fluid cells of patients with delayed cerebral ischemia following subarachnoid hemorrhage. Sci. Rep. 2021, 11, 16512. [Google Scholar] [CrossRef]
- Nakata, M.; Nakagomi, T.; Maeda, M.; Nakano-Doi, A.; Momota, Y.; Matsuyama, T. Induction of Perivascular Neural Stem Cells and Possible Contribution to Neurogenesis Following Transient Brain Ischemia/Reperfusion Injury. Transl. Stroke Res. 2017, 8, 131–143. [Google Scholar] [CrossRef]
- Bae, Y.H.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E.; Choi, H.G.; Ryu, H.; Hur, E.M.; Bu, Y.; et al. Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 2018, 9, 1125. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb. Cortex. 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Tyagi, A.; Kamal, M.A.; Poddar, N.K. Integrated Pathways of COX-2 and mTOR: Roles in Cell Sensing and Alzheimer’s Disease. Front. Neurosci. 2020, 14, 693. [Google Scholar] [CrossRef]
- Moussa, N.; Dayoub, N. Exploring the role of COX-2 in Alzheimer’s disease: Potential therapeutic implications of COX-2 inhibitors. Saudi Pharm. J. 2023, 31, 101729. [Google Scholar] [CrossRef]
- Nagano, T.; Tsuda, N.; Fujimura, K.; Ikezawa, Y.; Higashi, Y.; Kimura, S.H. Prostaglandin E(2) increases the expression of cyclooxygenase-2 in cultured rat microglia. J. Neuroimmunol. 2021, 361, 577724. [Google Scholar] [CrossRef]
- Guan, P.P.; Yu, X.; Zou, Y.H.; Wang, P. Cyclooxygenase-2 is critical for the propagation of β-amyloid protein and reducing the glycosylation of tau in Alzheimer’s disease. Cell Mol. Immunol. 2019, 16, 892–894. [Google Scholar] [CrossRef]
- Gopez, J.J.; Yue, H.; Vasudevan, R.; Malik, A.S.; Fogelsanger, L.N.; Lewis, S.; Panikashvili, D.; Shohami, E.; Jansen, S.A.; Narayan, R.K.; et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery 2005, 56, 590–604. [Google Scholar] [CrossRef]
- Fang, L.; Shen, S.; Liu, Q.; Liu, Z.; Zhao, J. Combination of NSAIDs with donepezil as multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2022, 75, 128976. [Google Scholar] [CrossRef]
- Goschorska, M.; Baranowska-Bosiacka, I.; Gutowska, I.; Tarnowski, M.; Piotrowska, K.; Metryka, E.; Safranow, K.; Chlubek, D. Effect of acetylcholinesterase inhibitors donepezil and rivastigmine on the activity and expression of cyclooxygenases in a model of the inflammatory action of fluoride on macrophages obtained from THP-1 monocytes. Toxicology 2018, 406–407, 9–20. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H. Autophagy in Traumatic Brain Injury: A New Target for Therapeutic Intervention. Front. Mol. Neurosci. 2018, 11, 190. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, W.; Lu, Y.; Zheng, Y.; Pan, L.; Wu, X.; Yuan, Y.; Shen, Z.; Ma, S.; Zhang, X.; et al. BNIP3L/NIX-mediated mitophagy: Molecular mechanisms and implications for human disease. Cell Death Dis. 2021, 13, 14. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, X.; Shan, H.; Zhang, M. Mitophagy in Traumatic Brain Injury: A New Target for Therapeutic Intervention. Oxid. Med. Cell. Longev. 2022, 2022, 4906434. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid. Redox Signal. 2015, 23, 565–577. [Google Scholar] [CrossRef]
- Khuanjing, T.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Anomasiri, A.; Chattipakorn, S.C.; Chattipakorn, N. Donepezil attenuated cardiac ischemia/reperfusion injury through balancing mitochondrial dynamics, mitophagy, and autophagy. Transl. Res. 2021, 230, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Hyder, A.A.; Wunderlich, C.A.; Puvanachandra, P.; Gururaj, G.; Kobusingye, O.C. The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 2007, 22, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; McCormick, W.C.; Kagan, S.H. Traumatic brain injury in older adults: Epidemiology, outcomes, and future implications. J. Am. Geriatr. Soc. 2006, 54, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.S.; Roebuck-Spencer, T.M.; Cernich, A. Cognitive changes and dementia risk after traumatic brain injury: Implications for aging military personnel. Alzheimers Dement. 2014, 10, S174–S187. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.S.; Sunyer, B.; Höger, H.; Lubec, G. Evaluation of spatial memory of C57BL/6J and CD1 mice in the Barnes maze, the Multiple T-maze and in the Morris water maze. Behav. Brain Res. 2009, 198, 58–68. [Google Scholar] [CrossRef]
- Baratz, R.; Tweedie, D.; Wang, J.Y.; Rubovitch, V.; Luo, W.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflamm. 2015, 12, 45. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youn, D.H.; Lee, Y.; Han, S.W.; Kim, J.-T.; Jung, H.; Han, G.S.; Yoon, J.I.; Lee, J.J.; Jeon, J.P. Therapeutic Effect of Donepezil on Neuroinflammation and Cognitive Impairment after Moderate Traumatic Brain Injury. Life 2024, 14, 839. https://doi.org/10.3390/life14070839
Youn DH, Lee Y, Han SW, Kim J-T, Jung H, Han GS, Yoon JI, Lee JJ, Jeon JP. Therapeutic Effect of Donepezil on Neuroinflammation and Cognitive Impairment after Moderate Traumatic Brain Injury. Life. 2024; 14(7):839. https://doi.org/10.3390/life14070839
Chicago/Turabian StyleYoun, Dong Hyuk, Younghyurk Lee, Sung Woo Han, Jong-Tae Kim, Harry Jung, Gui Seung Han, Jung In Yoon, Jae Jun Lee, and Jin Pyeong Jeon. 2024. "Therapeutic Effect of Donepezil on Neuroinflammation and Cognitive Impairment after Moderate Traumatic Brain Injury" Life 14, no. 7: 839. https://doi.org/10.3390/life14070839
APA StyleYoun, D. H., Lee, Y., Han, S. W., Kim, J.-T., Jung, H., Han, G. S., Yoon, J. I., Lee, J. J., & Jeon, J. P. (2024). Therapeutic Effect of Donepezil on Neuroinflammation and Cognitive Impairment after Moderate Traumatic Brain Injury. Life, 14(7), 839. https://doi.org/10.3390/life14070839