
Macrophage-Induced Pro-Fibrotic Gene Expression in Tubular Cells after Ischemia/Reperfusion Is Paralleled but Not Directly Mediated by C5a/C5aR1 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ischemia/Reperfusion Model in Rats
2.2. Immunohistochemical Staining of Rat Kidney Sections
2.3. In Situ Hybridization Combined with Immunofluorescence Staining
2.4. Human Cell Culture
2.5. Differentiation of THP-1 Monocytes to Macrophages
2.6. Flow Cytometry
2.7. Hypoxia and Macrophage/HPTC Co-Culture
2.8. Multiplex mRNA Expression Analysis
2.9. TGF-ß ELISA
2.10. C5a ELISA
2.11. Statistical Evaluation
3. Results
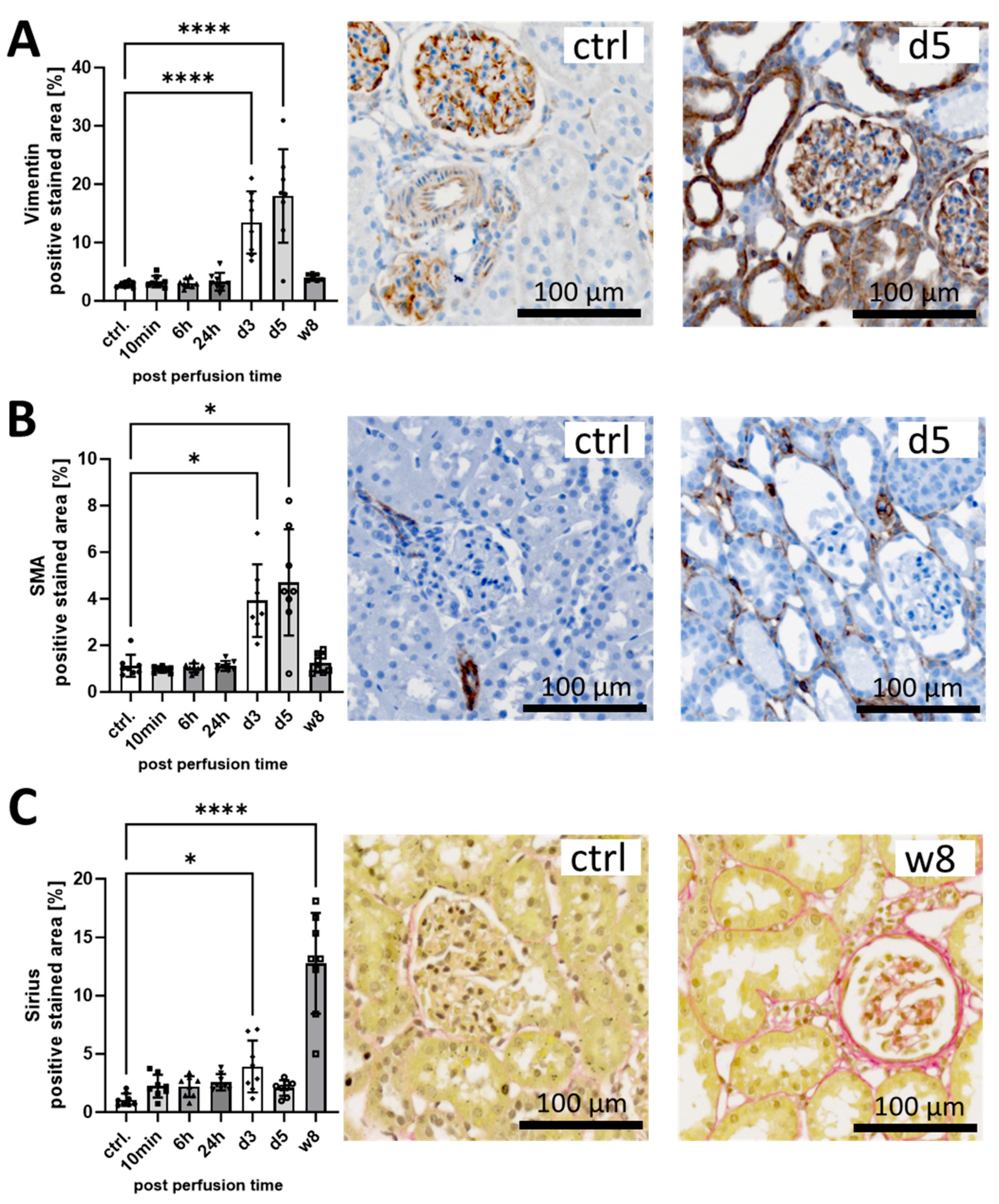
3.1. I/R Induces Early Pro-Fibrotic Response in Rat Kidneys and Leads to Long-Term Fibrotic Remodeling
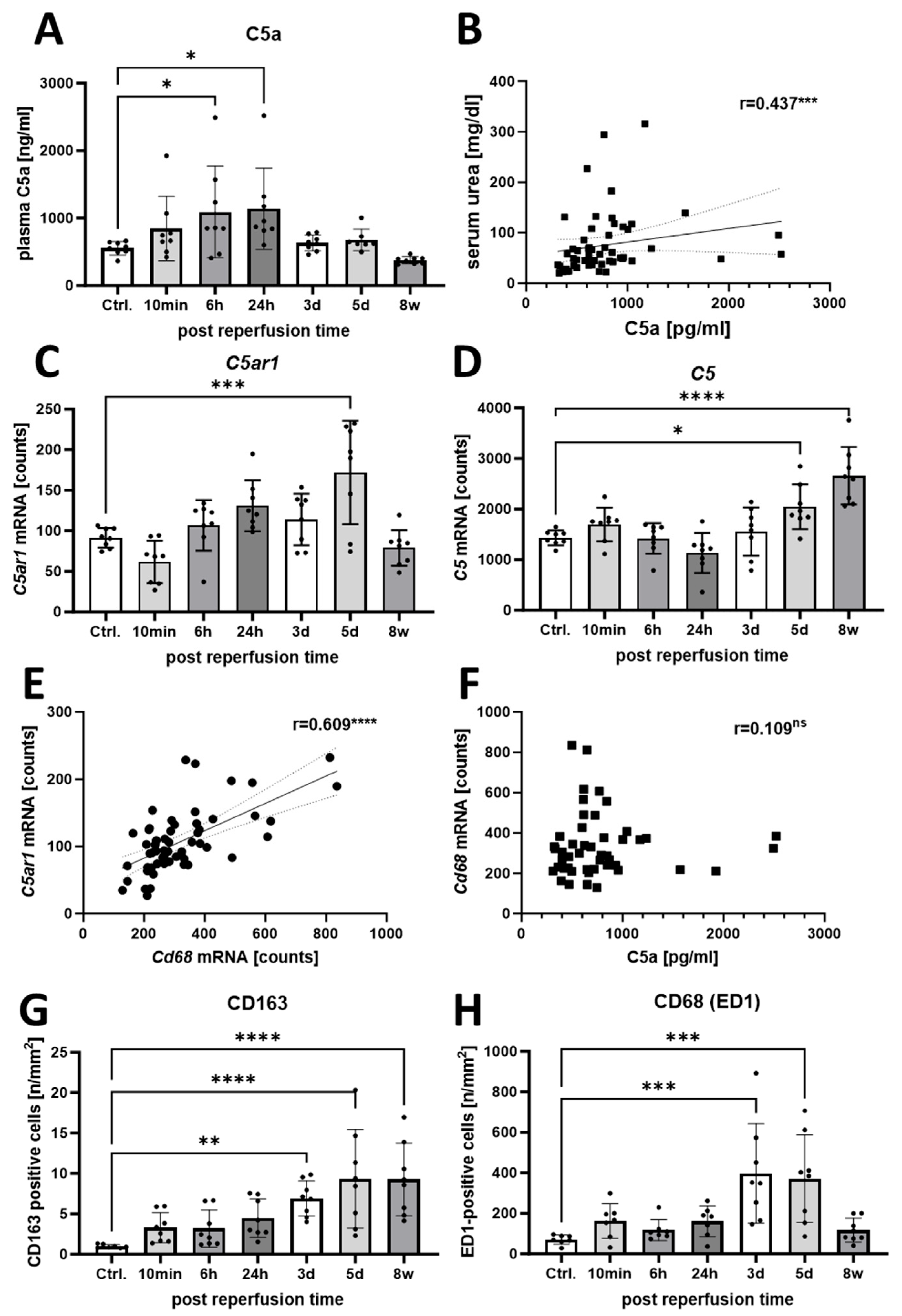
3.2. Increased Activation of the C5a/C5aR1 Axis Indicates a Possible Pathophysiological Role of C5a in the Rat I/R Model
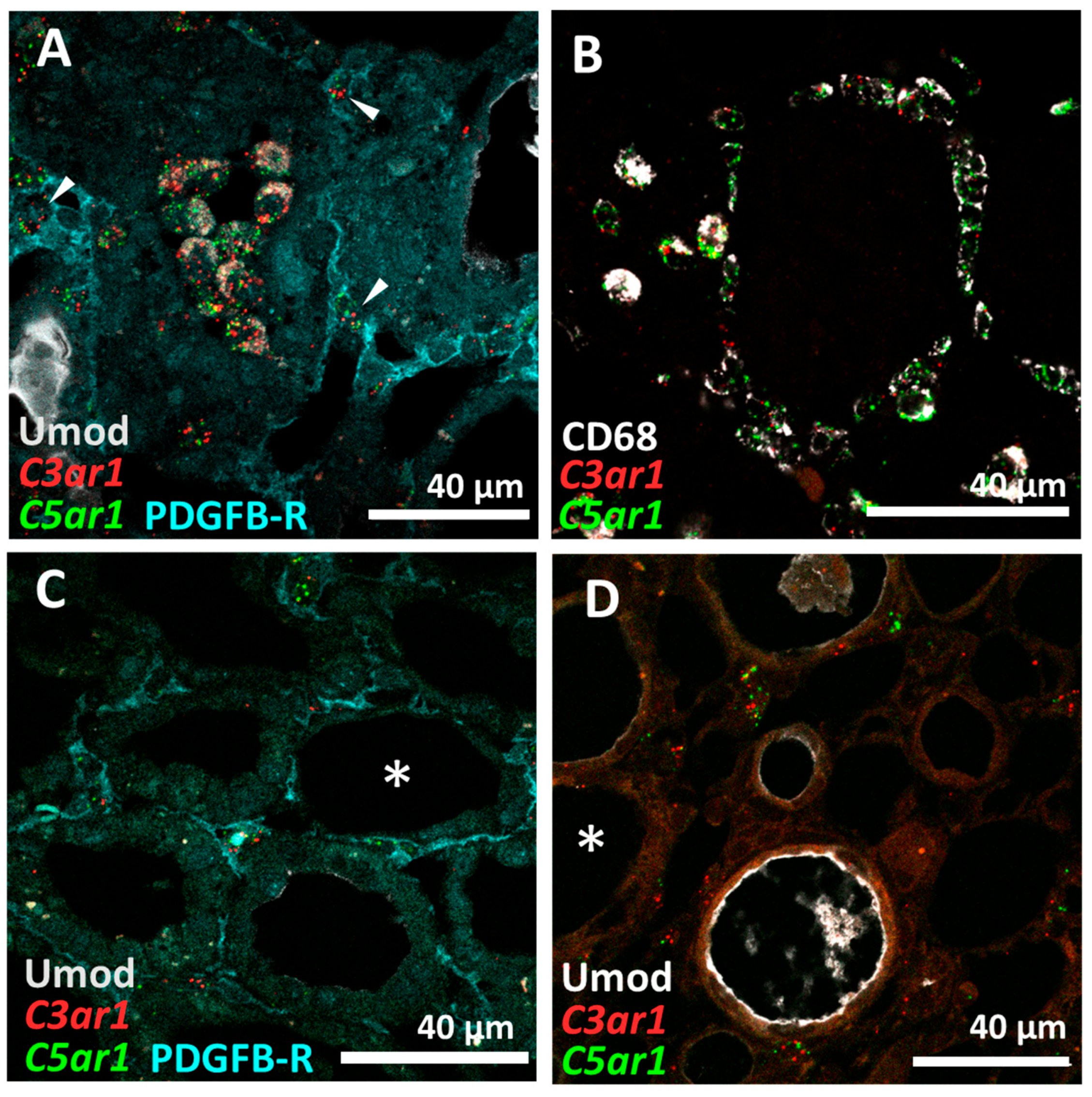
3.3. C5aR1 Is Expressed on CD68+ Macrophages but Not on Tubular Cells in the Rat I/R Model
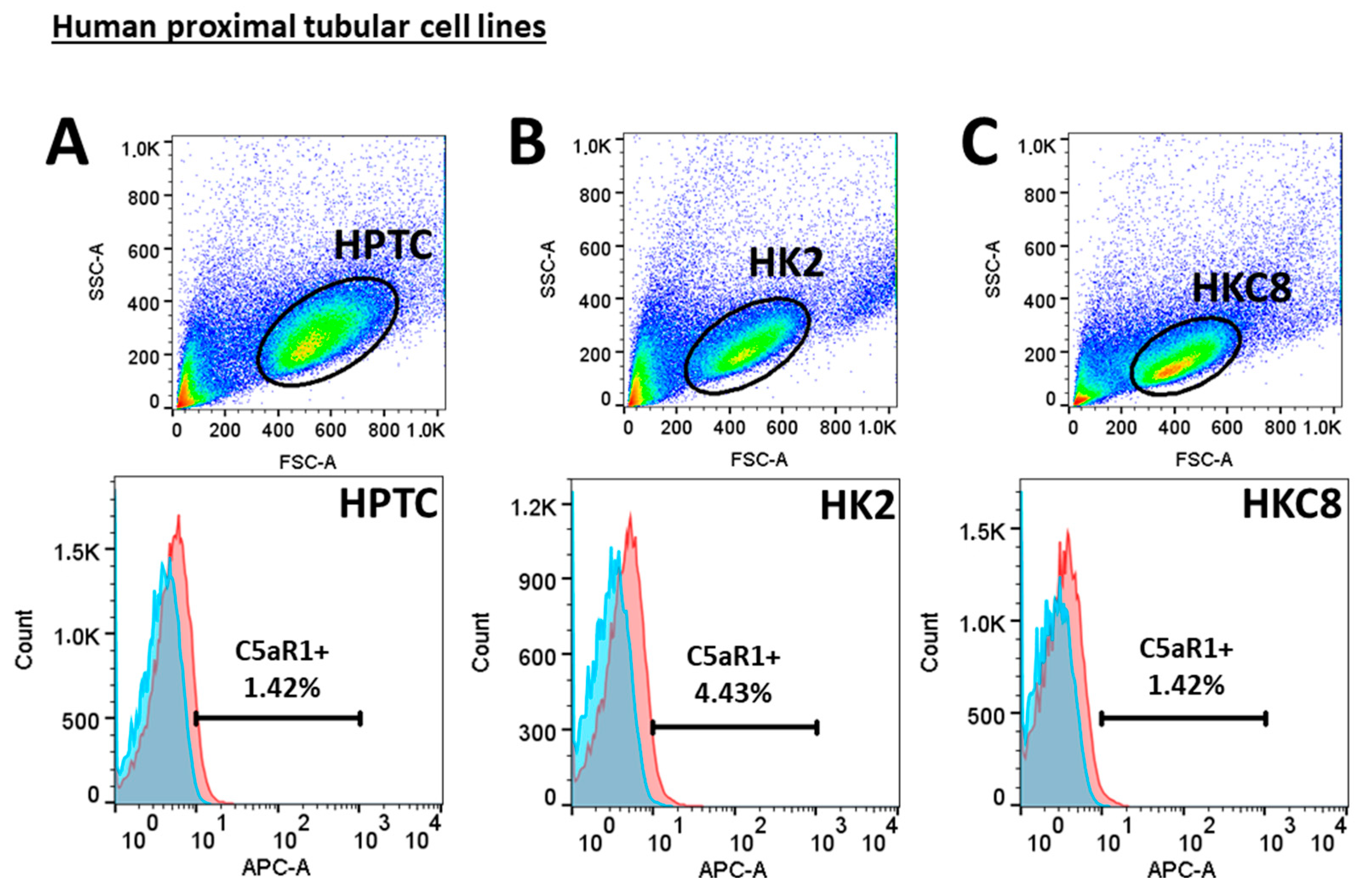
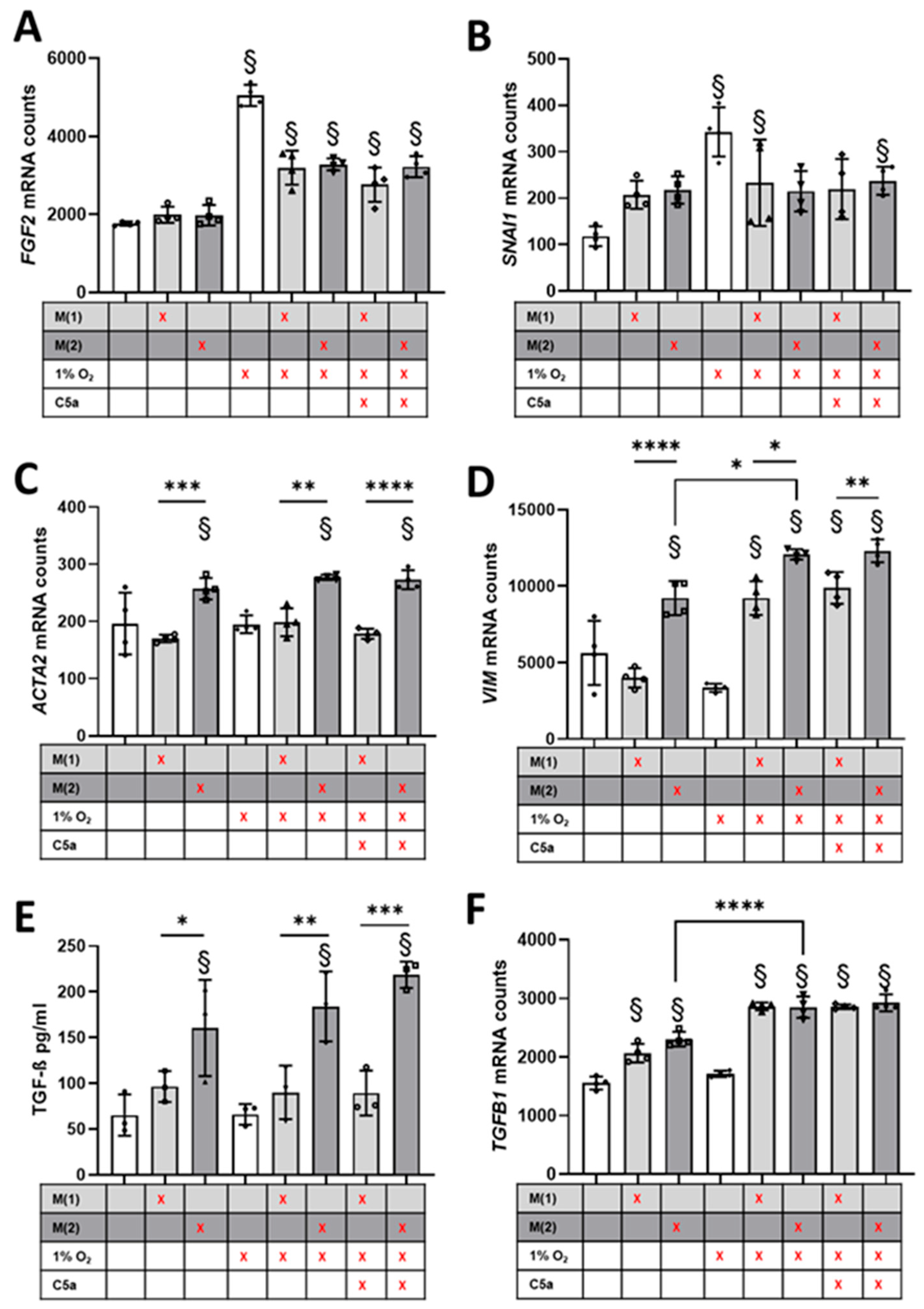
3.4. Crosstalk between Human Proximal Tubular Cells and Macrophages Significantly Induced Pro-Fibrotic Response but Was C5a-Independent
4. Discussion
4.1. Early Pro-Fibrotic Response in I/R Injury
4.2. The Role of C5a in I/R Injury and Fibrosis Induction
4.3. M2-like Macrophages Increase Pro-Fibrotic Response in Proximal Tubular Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danobeitia, J.S.; Djamali, A.; Fernandez, L.A. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair. 2014, 7, 16. [Google Scholar] [CrossRef]
- Danobeitia, J.S.; Ziemelis, M.; Ma, X.; Zitur, L.J.; Zens, T.; Chlebeck, P.J.; Van Amersfoort, E.S.; Fernandez, L.A. Complement inhibition attenuates acute kidney injury after ischemia-reperfusion and limits progression to renal fibrosis in mice. PLoS ONE 2017, 12, e0183701. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Saritas, T.; Kramann, R. Kidney Allograft Fibrosis: Diagnostic and Therapeutic Strategies. Transplantation 2021, 105, e114–e130. [Google Scholar] [CrossRef]
- Serón, D.; Moreso, F.; Ramón, J.M.; Hueso, M.; Condom, E.; Fulladosa, X.; Bover, J.; Gil-Vernet, S.; Castelao, A.M.; Alsina, J.; et al. Protocol renal allograft biopsies and the design of clinical trials aimed to prevent or treat chronic allograft nephropathy. Transplantation 2000, 69, 1849–1855. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.; Floege, J. Renal allograft fibrosis: Biology and therapeutic targets. Am. J. Transplant. 2015, 15, 863–886. [Google Scholar] [CrossRef]
- Yuan, Q.; Tan, R.J.; Liu, Y. Myofibroblast in Kidney Fibrosis: Origin, Activation, and Regulation. Adv. Exp. Med. Biol. 2019, 1165, 253–283. [Google Scholar]
- Mathern, D.R.; Heeger, P.S. Molecules Great and Small: The Complement System. Clin. J. Am. Soc. Nephrol. 2015, 10, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- Fayyazi, A.; Scheel, O.; Werfel, T.; Schweyer, S.; Oppermann, M.; Götze, O.; Radzun, H.J.; Zwirner, J. The C5a receptor is expressed in normal renal proximal tubular but not in normal pulmonary or hepatic epithelial cells. Immunology 2000, 99, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Nürge, B.; Schulz, A.L.; Kaemmerer, D.; Sänger, J.; Evert, K.; Schulz, S.; Lupp, A. Immunohistochemical identification of complement peptide C5a receptor 1 (C5aR1) in non-neoplastic and neoplastic human tissues. PLoS ONE 2021, 16, e0246939. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Li, K.; Smyth, L.A.; Xing, G.; Wang, N.; Meader, L.; Lu, B.; Sacks, S.H.; Zhou, W. C3a and C5a promote renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2012, 23, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Sacks, S.H.; Zhou, W. The role of complement in the early immune response to transplantation. Nat. Rev. Immunol. 2012, 12, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; D’Amati, A.; Crocetto, F.; Pandolfo, S.D.; Barone, B.; Ferro, M.; Spilotros, M.; Battaglia, M.; et al. Ischemia-Reperfusion Injury in Kidney Transplantation: Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 4332. [Google Scholar] [CrossRef] [PubMed]
- Vonbrunn, E.; Büttner-Herold, M.; Amann, K.; Daniel, C. Complement Inhibition in Kidney Transplantation: Where Are We Now? BioDrugs 2023, 37, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Franzin, R.; Stasi, A.; Fiorentino, M.; Stallone, G.; Cantaluppi, V.; Gesualdo, L.; Castellano, G. Inflammaging and Complement System: A Link between Acute Kidney Injury and Chronic Graft Damage. Front. Immunol. 2020, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Hertig, A.; Anglicheau, D.; Verine, J.; Pallet, N.; Touzot, M.; Ancel, P.Y.; Mesnard, L.; Brousse, N.; Baugey, E.; Glotz, D.; et al. Early epithelial phenotypic changes predict graft fibrosis. J. Am. Soc. Nephrol. 2008, 19, 1584–1591. [Google Scholar] [CrossRef]
- Kers, J.; Xu-Dubois, Y.C.; Rondeau, E.; Claessen, N.; Idu, M.M.; Roelofs, J.J.; Bemelman, F.J.; ten Berge, I.J.; Florquin, S. Intragraft tubular vimentin and CD44 expression correlate with long-term renal allograft function and interstitial fibrosis and tubular atrophy. Transplantation 2010, 90, 502–509. [Google Scholar] [CrossRef] [PubMed]
- de Matos, A.C.; Câmara, N.O.; Tonato, E.J.; Durão Júnior Mde, S.; Franco, M.F.; Moura, L.A.; Pacheco-Silva, A. Vimentin expression and myofibroblast infiltration are early markers of renal dysfunction in kidney transplantation: An early stage of chronic allograft dysfunction? Transplant. Proc. 2010, 42, 3482–3488. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Divanyan, A.; Jourd’heuil, F.L.; Goldman, R.D.; Ridge, K.M.; Jourd’heuil, D.; Lopez-Soler, R.I. Vimentin expression is required for the development of EMT-related renal fibrosis following unilateral ureteral obstruction in mice. Am. J. Physiol. Renal Physiol. 2018, 315, F769–F780. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef]
- Choudhry, N.; Li, K.; Zhang, T.; Wu, K.Y.; Song, Y.; Farrar, C.A.; Wang, N.; Liu, C.F.; Peng, Q.; Wu, W.; et al. The complement factor 5a receptor 1 has a pathogenic role in chronic inflammation and renal fibrosis in a murine model of chronic pyelonephritis. Kidney Int. 2016, 90, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Wu, W.; Wu, K.Y.; Cao, B.; Qiang, C.; Li, K.; Sacks, S.H.; Zhou, W. The C5a/C5aR1 axis promotes progression of renal tubulointerstitial fibrosis in a mouse model of renal ischemia/reperfusion injury. Kidney Int. 2019, 96, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kiafard, Z.; Tschernig, T.; Schweyer, S.; Bley, A.; Neumann, D.; Zwirner, J. Use of monoclonal antibodies to assess expression of anaphylatoxin receptors in tubular epithelial cells of human, murine and rat kidneys. Immunobiology 2007, 212, 129–139. [Google Scholar] [CrossRef]
- Yiu, W.H.; Li, R.X.; Wong, D.W.L.; Wu, H.J.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Sacks, S.H.; Zhou, W.; et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol. Dial. Transplant. 2018, 33, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Gou, R.; Huang, J.; Fu, P.; Chen, F.; Fan, W.-X.; Huang, Y.-Q.; Zang, L.; Wu, M.; Qiu, H.-Y.; et al. Effect of anaphylatoxin C3a, C5a on the tubular epithelial-myofibroblast transdifferentiation in vitro. Chin. Med. J. 2011, 124, 4039–4045. [Google Scholar]
- Fernandez, H.N.; Henson, P.M.; Otani, A.; Hugli, T.E. Chemotactic response to human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under stimulated in vivo conditions. J. Immunol. 1978, 120, 109–115. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Xu, J.; Xie, J.; Harris, D.C.H.; Zheng, G. The Role of Macrophages in Kidney Fibrosis. Front. Physiol. 2021, 12, 705838. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, H.; Zhang, D.; Zhang, Y.; Liu, B.; Wang, Y.; Zhou, H.; Xu, Z.X.; Wang, Y. The role of macrophages in fibrosis of chronic kidney disease. Biomed. Pharmacother. 2024, 177, 117079. [Google Scholar] [CrossRef]
- Pan, B.; Liu, G.; Jiang, Z.; Zheng, D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol. Biochem. 2015, 35, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.S. Transforming Growth Factor-β in the Acute Kidney Injury to Chronic Kidney Disease Transition. Nephron 2019, 143, 154–157. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Qi, R.; Wang, J.; Jiang, Y.; Qiu, Y.; Xu, M.; Rong, R.; Zhu, T. Snai1-induced partial epithelial-mesenchymal transition orchestrates p53-p21-mediated G2/M arrest in the progression of renal fibrosis via NF-κB-mediated inflammation. Cell Death Dis. 2021, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Zhuang, S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front. Physiol. 2020, 11, 569322. [Google Scholar] [CrossRef]
- Livingston, M.J.; Shu, S.; Fan, Y.; Li, Z.; Jiao, Q.; Yin, X.M.; Venkatachalam, M.A.; Dong, Z. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 2022, 19, 256–277. [Google Scholar] [CrossRef]
- Kierulf-Lassen, C.; Nielsen, P.M.; Qi, H.; Damgaard, M.; Laustsen, C.; Pedersen, M.; Krag, S.; Birn, H.; Nørregaard, R.; Jespersen, B. Unilateral nephrectomy diminishes ischemic acute kidney injury through enhanced perfusion and reduced pro-inflammatory and pro-fibrotic responses. PLoS ONE 2017, 12, e0190009. [Google Scholar] [CrossRef]
- Ransick, A.; Lindström, N.O.; Liu, J.; Zhu, Q.; Guo, J.J.; Alvarado, G.F.; Kim, A.D.; Black, H.G.; Kim, J.; McMahon, A.P. Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev. Cell 2019, 51, 399–413. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bleich, E.; Vonbrunn, E.; Büttner-Herold, M.; Amann, K.; Daniel, C. Macrophage-Induced Pro-Fibrotic Gene Expression in Tubular Cells after Ischemia/Reperfusion Is Paralleled but Not Directly Mediated by C5a/C5aR1 Signaling. Life 2024, 14, 1031. https://doi.org/10.3390/life14081031
Bleich E, Vonbrunn E, Büttner-Herold M, Amann K, Daniel C. Macrophage-Induced Pro-Fibrotic Gene Expression in Tubular Cells after Ischemia/Reperfusion Is Paralleled but Not Directly Mediated by C5a/C5aR1 Signaling. Life. 2024; 14(8):1031. https://doi.org/10.3390/life14081031
Chicago/Turabian StyleBleich, Erik, Eva Vonbrunn, Maike Büttner-Herold, Kerstin Amann, and Christoph Daniel. 2024. "Macrophage-Induced Pro-Fibrotic Gene Expression in Tubular Cells after Ischemia/Reperfusion Is Paralleled but Not Directly Mediated by C5a/C5aR1 Signaling" Life 14, no. 8: 1031. https://doi.org/10.3390/life14081031
APA StyleBleich, E., Vonbrunn, E., Büttner-Herold, M., Amann, K., & Daniel, C. (2024). Macrophage-Induced Pro-Fibrotic Gene Expression in Tubular Cells after Ischemia/Reperfusion Is Paralleled but Not Directly Mediated by C5a/C5aR1 Signaling. Life, 14(8), 1031. https://doi.org/10.3390/life14081031