
COVID-19 Coagulopathy

Abstract
:1. Introduction
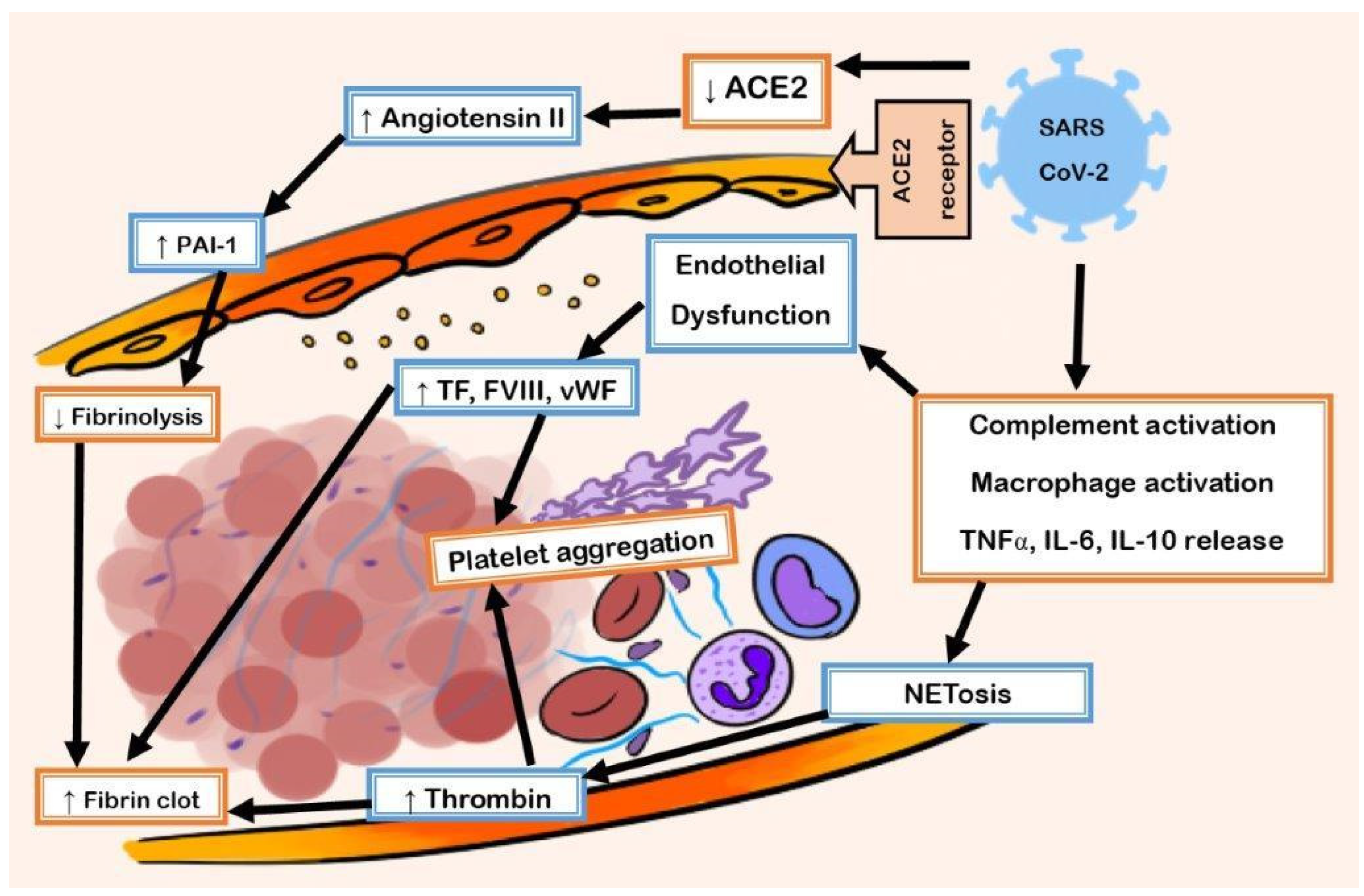
1.1. Pathophysiology of COVID-19 Coagulopathy
1.2. Entry of SARS-CoV-2 in ACE2
1.3. Hyperinflammatory Response and Oxidative Stress Damage
1.4. Prothrombotic Transformation of Endothelium
1.5. Role of the Complement System
1.6. Neutrophil Extracellular Traps (NETs)
1.7. Fibrinolysis Suppression
1.8. Endothelial Dysfunction
2. Biomarkers in COVID-19 Coagulopathy
3. Clinical Manifestations of COVID-19 Vasculopathy
3.1. Venous Thromboembolism
3.2. Arterial Thrombotic Events
3.3. Long COVID-19
4. Management of COVID-19 Related Venous Thromboembolism
4.1. Current Practice Guideline Recommendations for Prophylaxis of VTE in Hospitalized Patients with COVID-19
4.2. Recommendations for Prophylaxis of VTE in Non-Hospitalized Patients with COVID-19
4.3. Recommendations for Extended Duration Prophylaxis of VTE in Hospitalized Patients with COVID-19
4.4. Current Practice Guideline Recommendations for Treatment of VTE in Patients with COVID-19
Funding
Conflicts of Interest
References
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chang, X.N.; Pan, H.X.; Su, H.; Huang, B.; Yang, M. Pathological changes of the spleen in ten patients with coronavirus disease 2019 (COVID-19) by postmortem needle autopsy. Zhonghua Bing Li Xue Za Zhi 2020, 49, 576–582. [Google Scholar] [PubMed]
- Juthani, P.; Bhojwani, R.; Gupta, N. Coronavirus disease 2019 (COVID-19) manifestation as acute myocardial infarction in a young, healthy male. Case Rep Infect Dis. 2020, 2020, 8864985. [Google Scholar] [CrossRef] [PubMed]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P. Large-vessel stroke as a presenting feature of COVID-19 in the young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Hanff, T.C.; Mohareb, A.M.; Giri, J.; Cohen, J.B.; Chirinos, J.A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef]
- Ren, B.; Yan, F.; Deng, Z.; Zhang, S.; Xiao, L.; Wu, M.; Cai, L. Extremely high incidence of lower extremity deep venous thrombosis in 48 patients with severe COVID-19 in Wuhan. Circulation 2020, 142, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Lille ICU Haemostasis COVID-19 Group. Pulmonary embolism in patients with COVID-19: Awareness of an increased prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Aly, R.; Kaur, P.; Gupta, S.; Vasudev, R.; Virk, H.S. COVID-19 infection and arterial thrombosis: Report of three cases. Ann. Vasc. Surg. 2020, 70, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Choudry, F.A.; Hamshere, S.M.; Rathod, K.S.; Akhtar, M.M.; Archbold, R.A.; Guttmann, O.P. High thrombus burden in patients with COVID-19 presenting with ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2020, 76, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Fried, J.A.; Ramasubbu, K.; Bhatt, R.; Topkara, V.K.; Clerkin, K.J.; Horn, E. The variety of cardiovascular presentations of COVID-19. Circulation 2020, 141, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Tadros, A.; Hughes, D.P.; Dunmore, B.J.; Brindle, N.P. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood 2003, 102, 4407–4409. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Connors, J.M.; Warkentin, T.E.; Thachil, J.; Levi, M. The unique characteristics of COVID-19 coagulopathy. Crit. Care 2020, 24, 360. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased vWF and decreased ADAMTS-13 in COVID-19: Creating a milieu for (micro)thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418. [Google Scholar] [CrossRef] [PubMed]
- Englisch, C.N.; Tschernig, T.; Flockerzi, F.; Meier, C.; Bohle, R.M. Lesions in the lungs of fatal corona virus disease COVID-19. Ann. Anat. 2021, 234, 151657. [Google Scholar] [CrossRef] [PubMed]
- MacKman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-CoV2 associated thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jäckel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Mangoni, A.A. Serum Complement C3 and C4 and COVID-19 Severity and Mortality: A Systematic Review and Meta-Analysis with Meta-Regression. Front. Immunol. 2021, 12, 696085. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Martinod, K.; De Meyer, S.F. Neutrophil extracellular traps in arterial and venous thrombosis. Semin. Thromb. Hemost. 2019, 45, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Idell, S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit. Care Med. 2003, 31 (Suppl. S4), S213–S220. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J. Am. Coll. Surg. 2020, 231, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Khismatullin, R.R.; Ponomareva, A.A.; Nagaswami, C.; Ivaeva, R.A.; Montone, K.T.; Weisel, J.W.; Litvinov, R.I. Pathology of lung-specific thrombosis and inflammation in COVID-19. J. Thromb. Haemost. 2021, 19, 3062–3072. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, Y.; Ding, Y.; Luo, S.; Zheng, X.; Wu, X.; Liu, Z.; Ilyas, I.; Chen, S.; Han, S.; et al. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct. Target. Ther. 2021, 6, 266. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Muñoz Martin, A.J. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Scully, M. How I treat disseminated intravascular coagulation. Blood 2018, 131, 845–854. [Google Scholar] [CrossRef]
- Iba, T.; Warkentin, T.E.; Thachil, J.; Levi, M.; Levy, J.H. Proposal of the Definition for COVID-19-Associated Coagulopathy. J. Clin. Med. 2021, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Shah, K.; Patel, S.B.; Patel, F.S.; Osman, M.; Velagapudi, P.; Turagam, M.K.; Lakkireddy, D.; Garg, J. Elevated D-Dimer Levels Are Associated With Increased Risk of Mortality in Coronavirus Disease 2019: A Systematic Review and Meta-Analysis. Cardiol. Rev. 2020, 28, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef] [PubMed]
- Gerber, G.F.; Chaturvedi, S. How to recognize and manage COVID-19-associated coagulopathy. Hematol. Am. Soc. Hematol. Educ. Program. 2021, 2021, 614–620. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, F.; Chehade, S.; Lazo-Langner, A. Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb. Res. 2020, 192, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.E.; Kaatz, S.; Rosovsky, R.P.; Zon, R.L.; Pillai, S.; Robertson, W.E.; Elavalakanar, P.; Patell, R.; Khorana, A. COVID-19 and venous thromboembolism: A narrative review. Res. Pract. Thromb. Haemost. 2022, 6, e12666. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, O.H.; Daas, F.M.; Salunkhe, V.; Petrey, J.L.; Cosar, E.F.; Ramirez, J.; Akca, O. Is Microthrombosis the Main Pathology in Coronavirus Disease 2019 Severity?-A Systematic Review of the Postmortem Pathologic Findings. Crit. Care Explor. 2021, 3, e0427. [Google Scholar] [CrossRef] [PubMed]
- Sakr, Y.; Giovini, M.; Leone, M.; Pizzilli, G.; Kortgen, A.; Bauer, M.; Tonetti, T.; Duclos, G.; Zieleskiewicz, L.; Buschbeck, S.; et al. Pulmonary embolism in patients with coronavirus disease-2019 (COVID-19) pneumonia: A narrative review. Ann. Intensive Care 2020, 10, 124. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.-P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary embolism and deep vein thrombosis in COVID-19: A systematic review and meta-analysis. Radiology 2021, 298, E70–E80. [Google Scholar] [CrossRef] [PubMed]
- Malas, M.B.; Naazie, I.N.; Elsayed, N.; Mathlouthi, A.; Marmor, R.; Clary, B. Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: A systematic review and meta-analysis. EClinicalMedicine 2020, 29, 100639. [Google Scholar] [CrossRef] [PubMed]
- Gabet, A.; Grave, C.; Tuppin, P.; Olie, V.; Emmerich, J. One Year Prevalence of Venous Thromboembolism in Hospitalized COVID-19 Patients in France: Patients’ Characteristics, Time Trends, and Outcomes. Thromb. Haemost. 2022, 122, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Pasha, A.K.; McBane, R.D.; Chaudhary, R.; Padrnos, L.J.; Wysokinska, E.; Pruthi, R.; Ashrani, A.; Daniels, P.; Sridharan, M.; Wysokinski, W.E.; et al. Timing of venous thromboembolism diagnosis in hospitalized and non-hospitalized patients with COVID-19. Thromb. Res. 2021, 207, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ishiwata, Y.; Aoki, R.; Iwasawa, T.; Hagiwara, E.; Ogura, T.; Utsunomiya, D. Imaging of COVID-19: An update of current evidences. Diagn. Interv. Imaging 2021, 102, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hong, V.; Sy, L.S.; Glenn, S.C.; Ryan, D.S.; Morrissette, K.L.; Nelson, J.C.; Hambidge, S.J.; Crane, B.; Zerbo, O.; et al. Changes in incidence rates of outcomes of interest in vaccine safety studies during the COVID-19 pandemic. Vaccine 2022, 40, 3150–3158. [Google Scholar] [CrossRef] [PubMed]
- Kashi, M.; Jacquin, A.; Dakhil, B.; Zaimi, R.; Mahé, E.; Tella, E.; Bagan, P. Severe arterial thrombosis associated with COVID-19 infection. Thromb. Res. 2020, 192, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Johnson, R. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.; Quiwa, J.C.; Pillai, A.; Onwu, C.; Tharayil, Z.J.; Gupta, R. Superior mesenteric artery thrombosis and acute intestinal ischemia as a consequence of COVID-19 infection. Am. J. Case Rep. 2020, 21, e925753-1. [Google Scholar] [CrossRef] [PubMed]
- Melazzini, F.; Reduzzi, M.; Quaglini, S.; Fumoso, F.; Lenti, M.V.; Di Sabatino, A. Diagnostic delay of pulmonary embolism in COVID-19 patients. Front. Med. 2021, 8, 637375. [Google Scholar] [CrossRef] [PubMed]
- Michelen, M.; Manoharan, L.; Elkheir, N.; Cheng, V.; Dagens, A.; Hastie, C.; O’Hara, M.; Suett, J.; Dahmash, D.; Bugaeva, P.; et al. Characterising long COVID: A living systematic review. BMJ Glob. Health 2021, 6, e005427. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fan, Y.; Alwalid, O.; Li, N.; Jia, X.; Yuan, M.; Li, Y.; Cao, Y.; Gu, J.; Wu, H.; et al. Six-Month follow-up chest CT findings after severe COVID-19 pneumonia. Radiology 2021, 299, E177–E186. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Chen, S.; Zhang, Y.; Dong, F.; Zhang, Z.; Hu, B.; Zhu, Z.; Li, F.; Wang, X.; Wang, Y.; et al. Diffusion capacity abnormalities for carbon monoxide in patients with COVID-19 at 3-month follow-up. Eur. Respir. J. 2021, 58, 2003677. [Google Scholar] [CrossRef] [PubMed]
- Chioh, F.W.; Fong, S.W.; Young, B.E.; Wu, K.X.; Siau, A.; Krishnan, S.; Chan, Y.H.; Carissimo, G.; Teo, L.L.; Gao, F.; et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. Elife 2021, 10, e64909. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Thomas, W.; Retter, A.; Besser, M.; MacDonald, S.; Breen, K.A.; Desborough, M.J.; Hunt, B.J. Updated hospital associated venous thromboembolism outcomes with 90-days follow-up after hospitalisation for severe COVID-19 in two UK critical care units. Thromb. Res. 2020, 196, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; Reynolds, H.R.; Kumar, A.; Turgeon, A.F.; et al. Activ-4a, REMAP-CAP, ATTACC investigators. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar] [PubMed]
- ATTACC, ACTIV-4a, and REMAP-CAP Investigators. Therapeutic anticoagulation with heparin in noncritically ill patients with COVID-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, P.; Talasaz, A.H.; Rashidi, F.; Sharif-Kashani, B.; Beigmohammadi, M.T.; Farrokhpour, M.; Sezavar, S.H.; Payandemehr, P.; Dabbagh, A.; Moghadam, K.G.; et al. Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality among Patients with COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. JAMA 2021, 325, 1620–1630. [Google Scholar] [PubMed]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients with COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.; Furtado, R.H.; Macedo, A.V.; Bronhara, B.; Damiani, L.P.; Barbosa, L.M.; de Aveiro Morata, J.; Ramacciotti, E.; de Aquino Martins, P.; de Oliveira, A.L.; et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): An open-label, multicentre, randomised, controlled trial. Lancet 2021, 397, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Brooks, M.M.; Sciurba, F.C.; Krishnan, J.A.; Bledsoe, J.R.; Kindzelski, A.; Baucom, A.L.; Kirwan, B.-A.; Eng, H.; Martin, D.; et al. Effect of Antithrombotic Therapy on Clinical Outcomes in Outpatients with Clinically Stable Symptomatic COVID-19: The ACTIV-4B Randomized Clinical Trial. JAMA 2021, 326, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Ramacciotti, E.; Agati, L.B.; Calderaro, D.; Aguiar, V.C.; Spyropoulos, A.C.; de Oliveira, C.C.; Dos Santos, J.L.; Volpiani, G.G.; Sobreira, M.L.; Joviliano, E.E.; et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): An open-label, multicentre, randomised, controlled trial. Lancet 2022, 399, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Burnett, A.; Allen, A.; Ansell, J.; Blumenstein, M.; Clark, N.P.; Crowther, M.; Dager, W.E.; Deitelzweig, S.B.; Ellsworth, S.; et al. Thromboembolic prevention and anticoagulant therapy during the COVID-19 pandemic: Updated clinical guidance from the anticoagulation forum. J. Thromb. Thrombolysis 2022, 54, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Bowles, L.; Platton, S.; Yartey, N.; Dave, M.; Lee, K.; Hart, D.P.; MacDonald, V.; Green, L.; Sivapalaratnam, S.; Pasi, K.J.; et al. Lupus Anticoagulant and Abnormal Coagulation Tests in Patients with COVID-19. N. Engl. J. Med. 2020, 383, 288–290. [Google Scholar] [CrossRef]

{kind=link}
| Proposed CAC Criteria: ≥2 of the following: | ISTH DIC Criteria: ≥5 Points | |
|---|---|---|
| D-dimer | >2x ULN | <5x ULN = 2, ≥5x ULN= 3 |
| Platelet count | <150 × 109/L | <100 = 1, <50 = 2 |
| Prolonged PT | >1 s or INR > 1.2 | ≥3 s to <6 s = 1, ≥6 s = 2 |
| Thrombosis | Presence, micro or macrovascular | Not part of the criteria |
| Fibrinogen | Not part of the criteria, but considered at risk for CAC if elevated | ≤1.0 g/L = 1 |
| Other risk factors: vWF > 2x ULN, presence of high-titer antiphospholipid antibodies and lupus anticoagulant | ||
| Society/Guideline |
Outpatient Prophylaxis | Non-Critically Ill Prophylaxis | Critically Ill Prophylaxis | Post Discharge Prophylaxis |
|---|---|---|---|---|
| ASH | No recommendation | Therapeutic dose | Prophylactic dose | Yes |
| ISTH | No recommendation | Prophylactic dose Or intermediate dose | Prophylactic dose Or intermediate dose | In high-risk patients |
| Chest 2020 | No recommendation | Prophylactic dose | Prophylactic dose | No recommendation |
| NIH | No recommendation | Therapeutic dose if elevated d-dimer and on low flow oxygen | Prophylactic dose | Routine use not recommended |
| Anticoagulation Forum | No recommendation | Prophylactic dose, consider therapeutic in high-risk patients | Prophylactic dose | In high-risk patients |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rettew, A.; Garrahy, I.; Rahimian, S.; Brown, R.; Sangha, N. COVID-19 Coagulopathy. Life 2024, 14, 953. https://doi.org/10.3390/life14080953
Rettew A, Garrahy I, Rahimian S, Brown R, Sangha N. COVID-19 Coagulopathy. Life. 2024; 14(8):953. https://doi.org/10.3390/life14080953
Chicago/Turabian StyleRettew, Andrew, Ian Garrahy, Shoja Rahimian, Rebecca Brown, and Navdeep Sangha. 2024. "COVID-19 Coagulopathy" Life 14, no. 8: 953. https://doi.org/10.3390/life14080953
APA StyleRettew, A., Garrahy, I., Rahimian, S., Brown, R., & Sangha, N. (2024). COVID-19 Coagulopathy. Life, 14(8), 953. https://doi.org/10.3390/life14080953