
Screening and Validation of Leaf Width-Related Genes in Inbred Maize Lines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Leaf Width Phenotypic Data Analysis
2.3. Whole-Genome Sequencing
2.4. GWAS
2.5. Candidate Gene Validation
3. Results
3.1. Study Population Phenotypic Characteristics
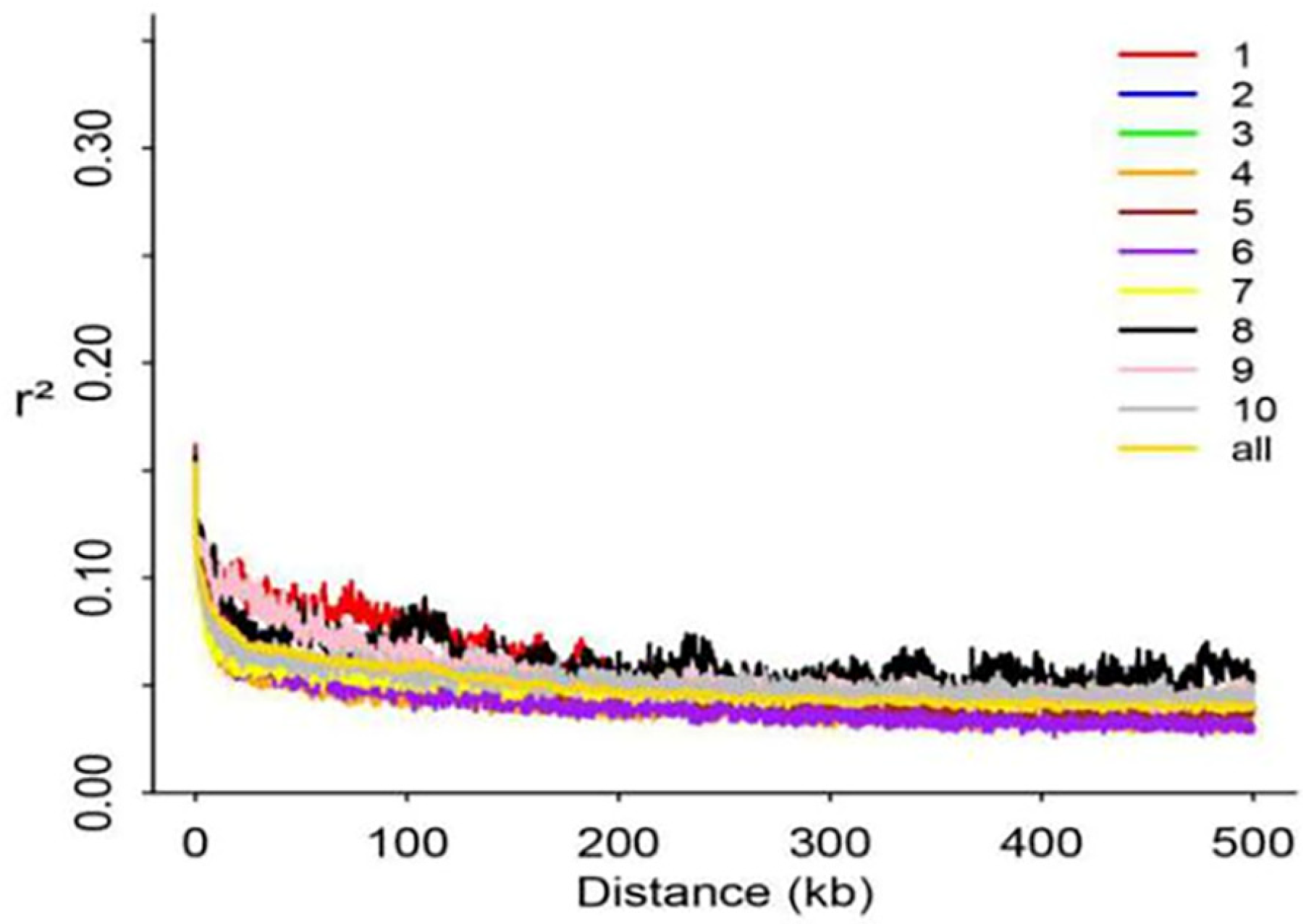
3.2. NGS Data Quality and LD Analysis
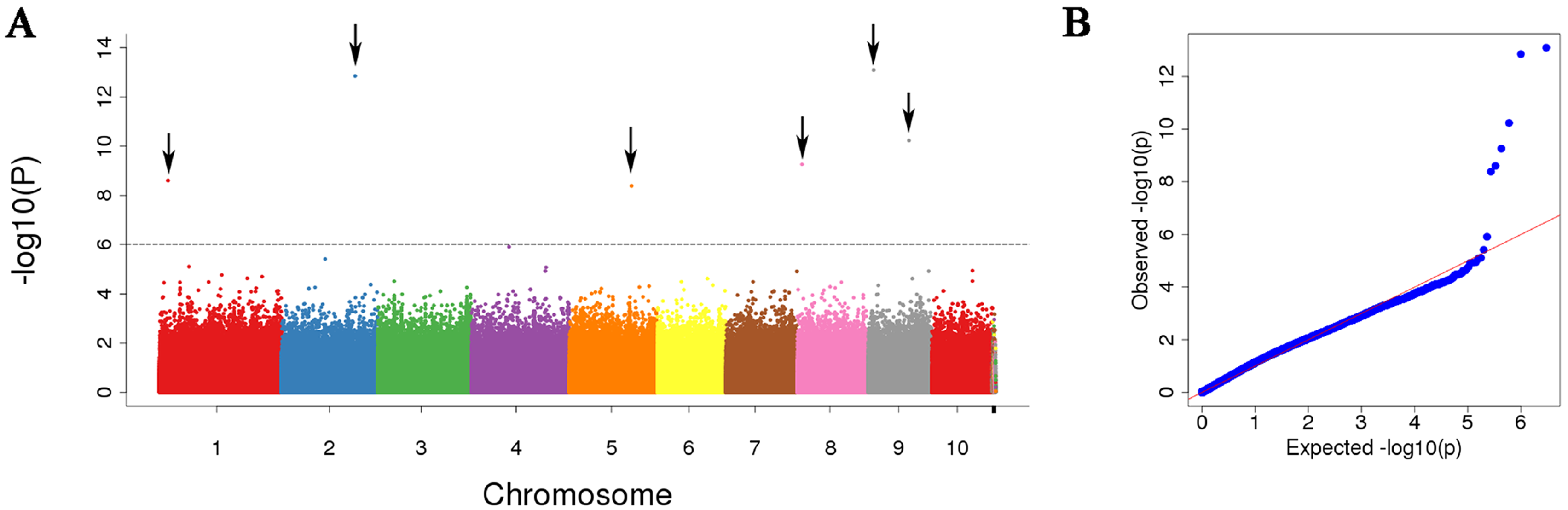
3.3. Leaf Width-Related SNP Identification
3.4. Identification and Functional Annotation of Leaf Width-Related Candidate Genes
3.5. Functional Confirmation of SNP-Associated Gene Variants
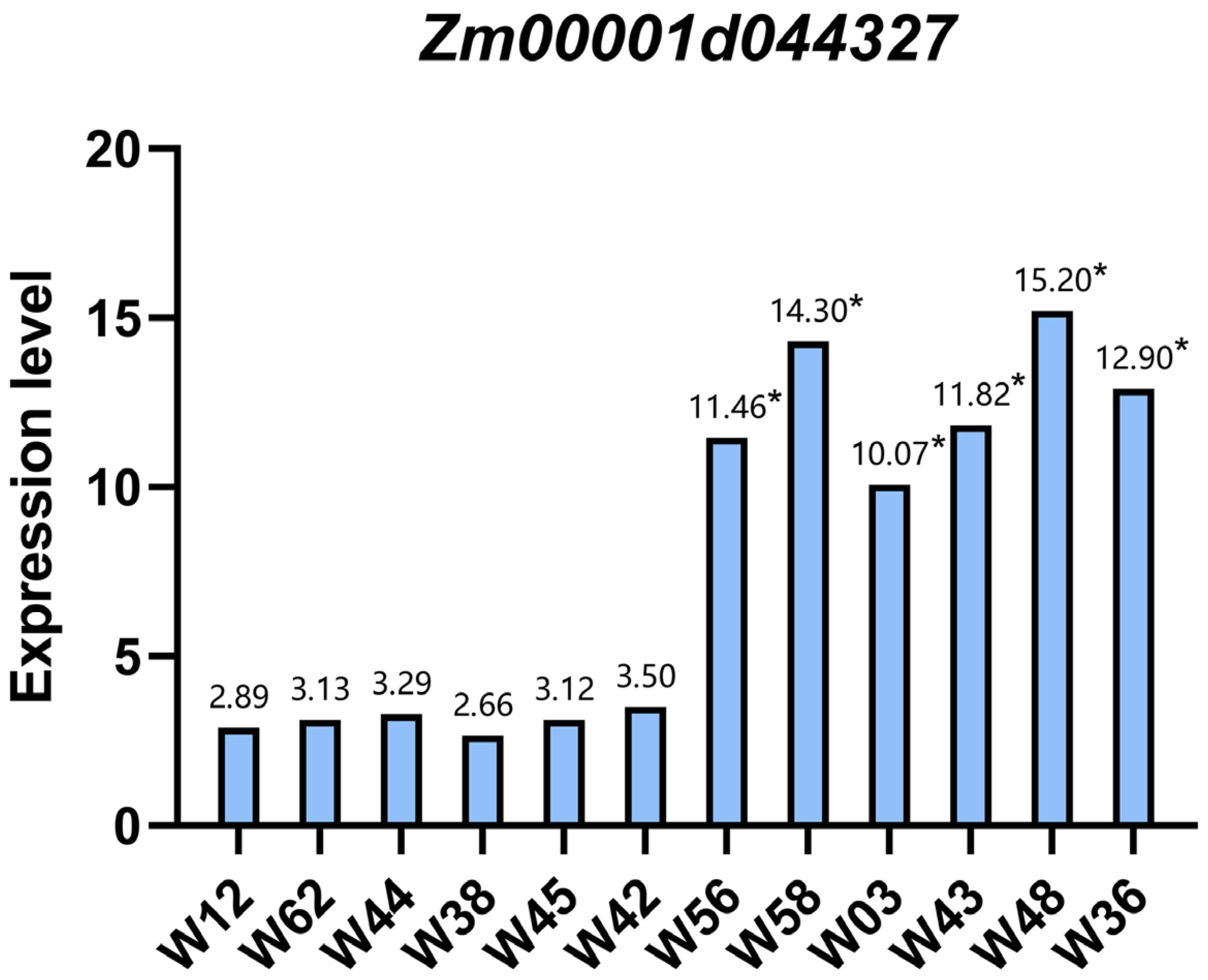
3.6. qPCR Analysis of the Zm00001d044327 Candidate Gene
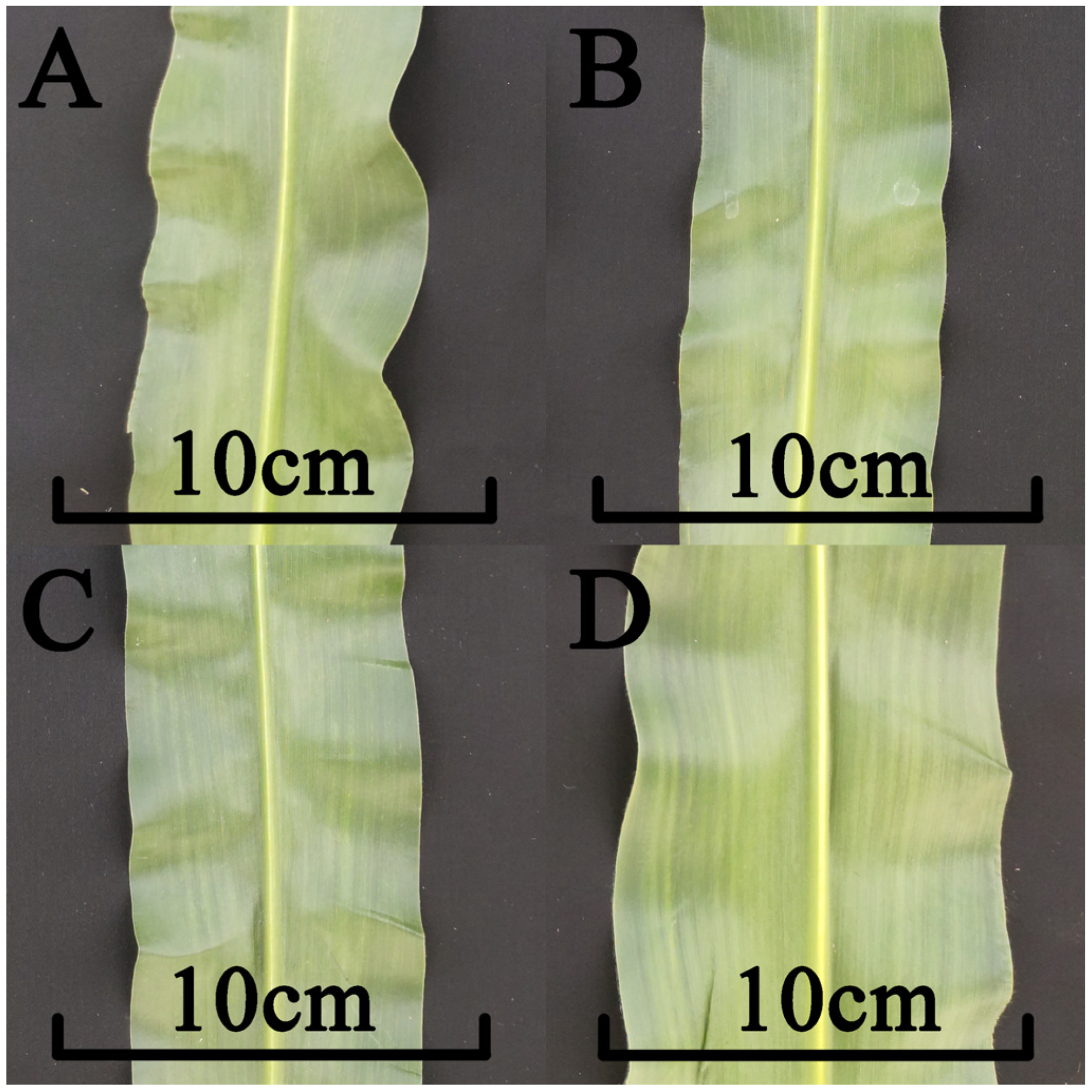
3.7. Assessment of the Phenotypic Impact of Zm00001d044327 Candidate Gene Mutations
4. Discussion
4.1. Leaf Width-Related SNP Markers
4.2. Candidate Gene Functional Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borba, A.R.; Serra, T.S.; Górska, A.; Gouveia, P.; Cordeiro, A.M.; Reyna-Llorens, I.; Kneřová, J.; Barros, P.M.; A Abreu, I.; Oliveira, M.M.; et al. Synergistic binding of bHLH transcription factors to the promoter of the maize NADP-ME gene used in C4 photosynthesis is based on an ancient code found in the ancestral C3 state. Mol. Biol. Evol. 2018, 35, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Czedik-Eysenberg, A.; Mertz, R.A.; Si, Y.; Tohge, T.; Nunes-Nesi, A.; Arrivault, S.; Dedow, L.K.; Bryant, D.W.; Zhou, W.; et al. Comparative analyses of C4 and C3 photosynthesis in developing leaves of maize and rice. Nat. Biotechnol. 2014, 32, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Hou, Y.-H.; Sun, R.; Zhu, P.; Dong, Z.-Q.; Zhao, M. Effects of Planting Density on Yield Performance and Density-Tolerance Analysis for Maize Hybrids. Acta Agron. Sin. 2010, 36, 1153–1160. [Google Scholar] [CrossRef]
- Gou, L.; Huang, J.J.; Sun, R. Variation characteristic of stalk penetration strength of maize with different density-tolerance varieties. Trans. CSAE 2010, 26, 156–162. [Google Scholar] [CrossRef]
- Mansfield, B.D. Survey of plant density tolerance in U.S. maize germplasm. Crop. Sci. 2012, 54, 157–173. [Google Scholar] [CrossRef]
- Simon, A.C.M.; Lopez-Nicora, H.D.; Lindsey, L.E.; Niblack, T.L.; Paul, P.A. Incidence, Population Density and Spatial Heterogeneity of Plant-Parasitic Nematodes in Corn Fields in Ohio. Plant Dis. 2018, 102, 2453–2464. [Google Scholar] [CrossRef]
- Liu, R.; Meng, Q.; Zheng, F.; Kong, L.; Yuan, J.; Lübberstedt, T. Genetic mapping of QTL for maize leaf width combining RIL and IF2 populations. PLoS ONE 2017, 12, e0189441. [Google Scholar] [CrossRef] [PubMed]
- Pelleschi, S.; Leonardi, A.; Rocher, J.P.; Cornic, G.; de Vienne, D.; Thévenot, C.; Prioul, J.L. Analysis of the relationships between growth, photosynthesis and carbohydrate metabolism using quantitative trait loci (QTLs) in young maize plants subjected to water deprivation. Mol. Breed. 2006, 17, 21–39. [Google Scholar] [CrossRef]
- Ku, L.X.; Zhang, J.; Guo, S.L.; Liu, H.Y.; Zhao, R.F.; Chen, Y.H. Integrated multiple population analysis of leaf architecture traits in maize (Zea mays L.). J. Exp. Bot. 2012, 63, 261–274. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, M.; Zhang, Z.; Wang, Z.; Wu, N.; Song, Y.; Wang, P. Screening and verification of genes associated with leaf angle and leaf orientation value in inbred maize lines. PLoS ONE 2018, 13, e0208386. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef]
- Nordborg, M.; Weigel, D. Next-generation genetics in plants. Nature 2008, 456, 720–723. [Google Scholar] [CrossRef]
- Gilad, Y.; Pritchard, J.K.; Thornton, K. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 2009, 25, 463–471. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The genetic architecture of maize flowering time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef]
- Wang, B.B.; Zhu, Y.B.; Zhu, J.J.; Liu, Z.; Liu, H.; Dong, X.; Guo, J.; Li, W.; Chen, J.; Gao, C.; et al. Identification and Fine-Mapping of a Major Maize Leaf Width QTL in a Re-sequenced Large Recombinant Inbred Lines Population. Front. Plant Sci. 2018, 9, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.L.; Li, C.X.; Gong, S.C.; Li, G.L.; Hu, G.H.; Wang, M.Q.; Yang, J.F. Genetic Correlation and Path Analysis of Yield and Agronomic Characteristics of Maize Hybrids in SPSS Software. Crops 2019, 3, 86–90. [Google Scholar] [CrossRef]
- Lu, S.; Li, M.; Zhang, M.; Lu, M.; Wang, X.; Wang, P.; Liu, W. Genome-Wide Association Study of Plant and Ear Height in Maize. Trop. Plant Biol. 2020, 13, 262–273. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Harper, L.C.; Schaeffer, M.L.; Thistle, J.; Gardiner, J.M.; Andorf, C.M.; Campbell, D.A.; Cannon, E.K.; Braun, B.L.; Birkett, S.M.; Lawrence, C.J.; et al. The maizeGDB genome browser tutorial: One example of database outreach to biologists via video. Database 2011, 2011, bar016. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, C.J.; Wang, J. Real-time fluorescent quantitative PCR and application in scientific research. Biotechnol. Bull. 2009, 2, 73–76. [Google Scholar] [CrossRef]
- Tang, D. QTL Mapping and Analysis for Leaf Width and Length in the Maize (Zea mays L.). Master’s Thesis, Sichuan Agricultural University, Sichuan, China, 2013. [Google Scholar]
- Zhang, Z. QTL Mapping and Analysis of Leaf Type Related Traits in Maize, 1st ed.; Sichuan Agricultural University: Sichuan, China, 2015. [Google Scholar]
- Ku, L. Studies on the Molecular Genetic Mechanism of Plant Architecture Traits in Maize (Zea mays L.). Ph.D. Thesis, He Nan Agricultural School, Zhengzhou, China, 2010. [Google Scholar]
- Mayaka, J.B.; Huang, Q.; Xiao, Y.; Zhong, Q.; Ni, J.; Shen, Y. The Lonely Guy (LOG) homologue SiRe_0427 from the thermophilic archaeon Sulfolobus islandicus REY15A is a phosphoribohydrolase representing a novel group. Appl. Environ. Microb. 2019, 85, e01739-19. [Google Scholar] [CrossRef]
- Ferrari, C.; Amaral, F.; Ferreira, J. Expressed Proteins of Herbaspirillum seropedicae in Maize (DKB240) Roots-Bacteria Interaction Revealed Using Proteomics. Appl. Biochem. Biotechnol. 2014, 174, 2267–2277. [Google Scholar] [CrossRef]
- Paulsen, H. Chlorophyll a/b-binding proteins. Photochem. Photobiol. 1995, 62, 367–382. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′-3′) | Candidate Gene |
|---|---|---|
| QLWS | TGCTCAGTCAGCAGCATATC | Zm00001d044327 |
| QLWAS | CCTTCCTTCCTCCCTACTTCTA |
| Year | SNP | Physical Position | Chr | Genotype | MAF | log10 (P) | Distance (bp) | Contribution (r2) | Candidate Gene | Functional Annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| 2015 | sLW-226131901 | 226131901 | 2 | A/G | 0.05 | 6.31 | −9706 | 6.20% | Zm00001d007267 | Chlorophyll a-b binding protein CP26, chloroplastic |
| sLW-225322565 | 225322565 | 3 | C/T | 0.07 | 6.33 | 6% | ||||
| sLW-225305520 | 225305520 | 3 | C/T | 0.30 | 6.35 | 0 | 13.20% | Zm00001d044327 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase LOG7 | |
| sLW-141511859 | 141511859 | 5 | G/A | 0.11 | 6.11 | 4.90% | ||||
| sLW-150065211 | 150065211 | 6 | T/A | 0.06 | 6.07 | 0.40% | ||||
| sLW-30006587 | 30006587 | 7 | T/G | 0.34 | 6.73 | 3.20% | ||||
| sLW-104304481 | 104304481 | 7 | C/A | 0.05 | 6.17 | 3128 | 7.10% | Zm00001d020285 | Putative pumilio homolog 7, chloroplastic | |
| sLW-104304481 | 104304481 | 7 | C/A | 0.05 | 6.17 | −2528 | 7.10% | Zm00001d020287 | hypothetical protein ZEAMMB73 | |
| sLW-68332493 | 68332493 | 8 | C/T | 0.12 | 6.04 | 1.90% | ||||
| 2016 | sLW-20566973 | 20566973 | 1 | G/T | 0.07 | 8.60 | 3.10% | |||
| sLW-186867035 | 186867035 | 2 | C/A | 0.07 | 12.81 | 2768 | 13.60% | Zm00001d005758 | ||
| sLW-153643898 | 153643898 | 5 | G/T | 0.13 | 8.38 | 14.20% | ||||
| sLW-11019663 | 11019663 | 8 | C/T | 0.49 | 9.26 | 7.20% | ||||
| sLW-12911742 | 12911742 | 9 | T/A | 0.10 | 13.09 | 3.70% | ||||
| sLW-100159475 | 100159475 | 9 | T/A | 0.13 | 10.23 | 11.60% |
| Mutant | Leaf Width above the Uppermost Ear (cm) | Sig. |
|---|---|---|
| wild type (B73) | 9.8 | A |
| ZMLW-1 | 7.2 | B |
| ZMLW-2 | 6.8 | B |
| ZMLW-3 | 7.4 | B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, S.; Wang, Q.; Yin, J.; Zheng, S.; Gao, T.; Zhou, X.; Zhang, J.; Xing, Y.; Ma, Y.; Wang, M.; et al. Screening and Validation of Leaf Width-Related Genes in Inbred Maize Lines. Life 2024, 14, 1057. https://doi.org/10.3390/life14091057
Lu S, Wang Q, Yin J, Zheng S, Gao T, Zhou X, Zhang J, Xing Y, Ma Y, Wang M, et al. Screening and Validation of Leaf Width-Related Genes in Inbred Maize Lines. Life. 2024; 14(9):1057. https://doi.org/10.3390/life14091057
Chicago/Turabian StyleLu, Shi, Qi Wang, Junqi Yin, Shubo Zheng, Tingting Gao, Xudong Zhou, Jianxin Zhang, Yuexian Xing, Yingjie Ma, Min Wang, and et al. 2024. "Screening and Validation of Leaf Width-Related Genes in Inbred Maize Lines" Life 14, no. 9: 1057. https://doi.org/10.3390/life14091057
APA StyleLu, S., Wang, Q., Yin, J., Zheng, S., Gao, T., Zhou, X., Zhang, J., Xing, Y., Ma, Y., Wang, M., Zhou, D., Lu, M., Liu, W., Wang, P., & Zhang, Z. (2024). Screening and Validation of Leaf Width-Related Genes in Inbred Maize Lines. Life, 14(9), 1057. https://doi.org/10.3390/life14091057