
Conformational Dynamics of Mitochondrial Inorganic Pyrophosphatase hPPA2 and Its Changes Caused by Pathogenic Mutations


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Analysis of the Possible Effects of Pathogenic Mutations on hPPA2 Structure
3.2. Conformational Dynamics of hPPA2 Studied by Molecular Dynamics Simulation
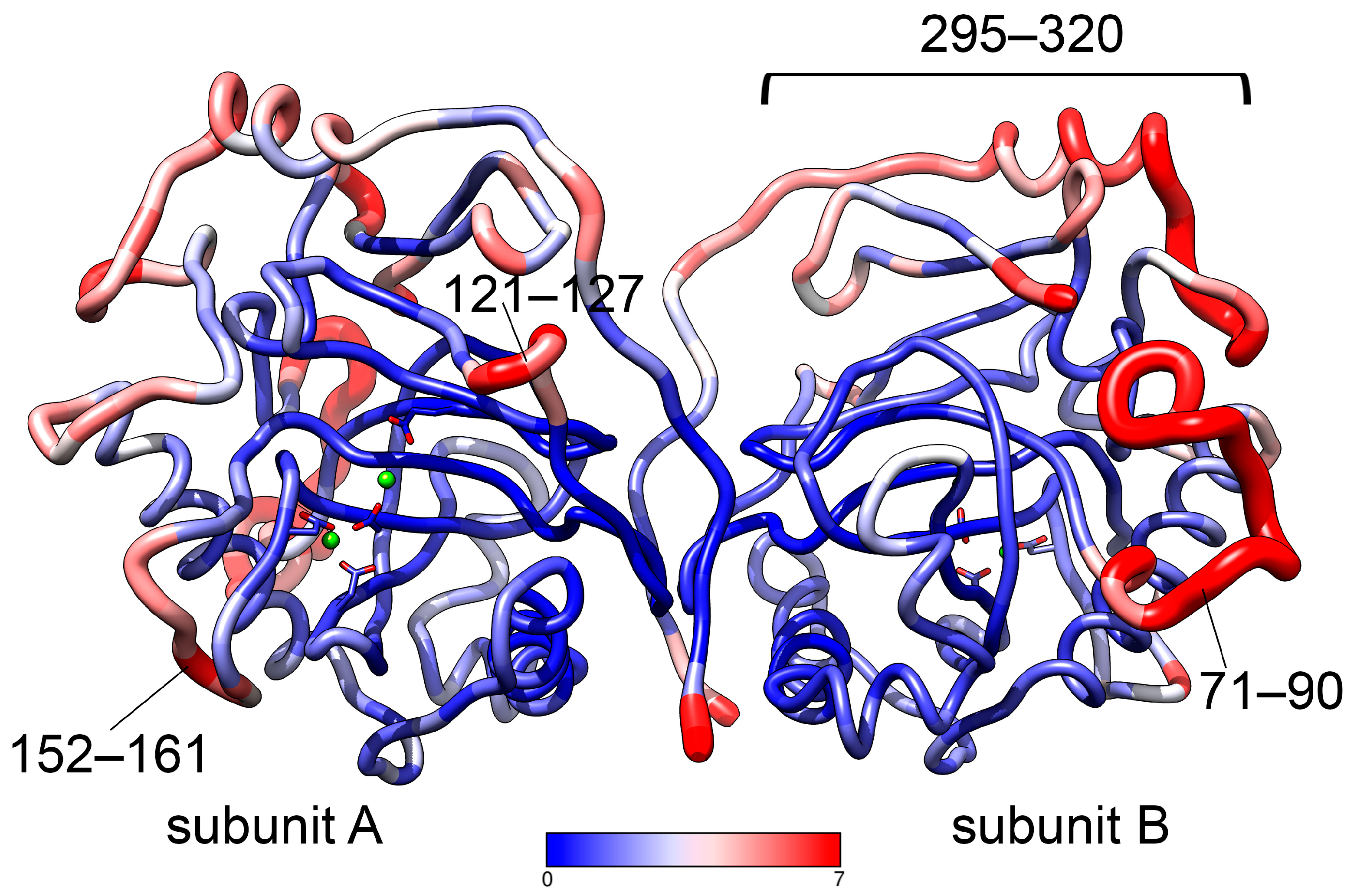
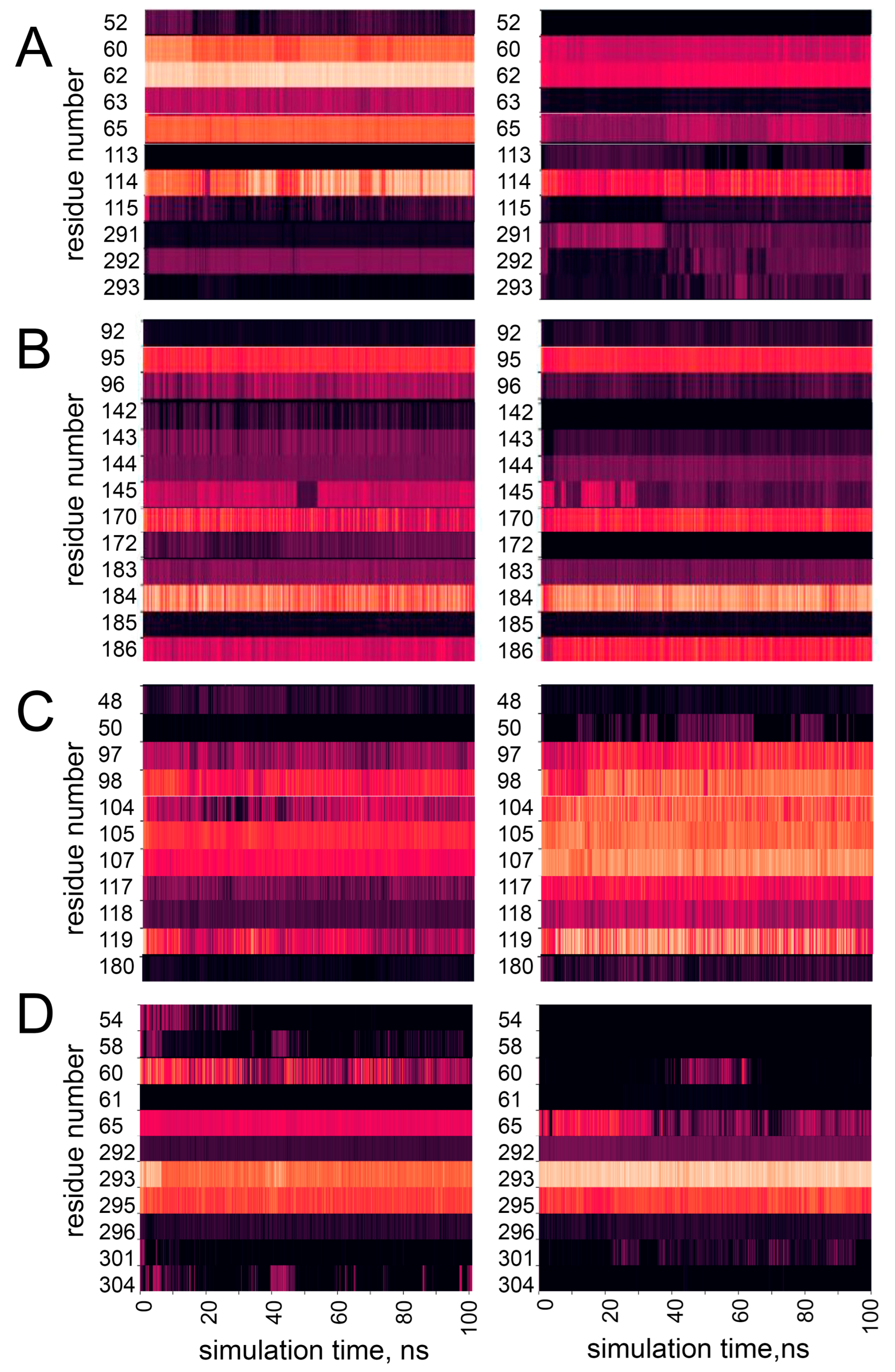
3.2.1. General Characteristics
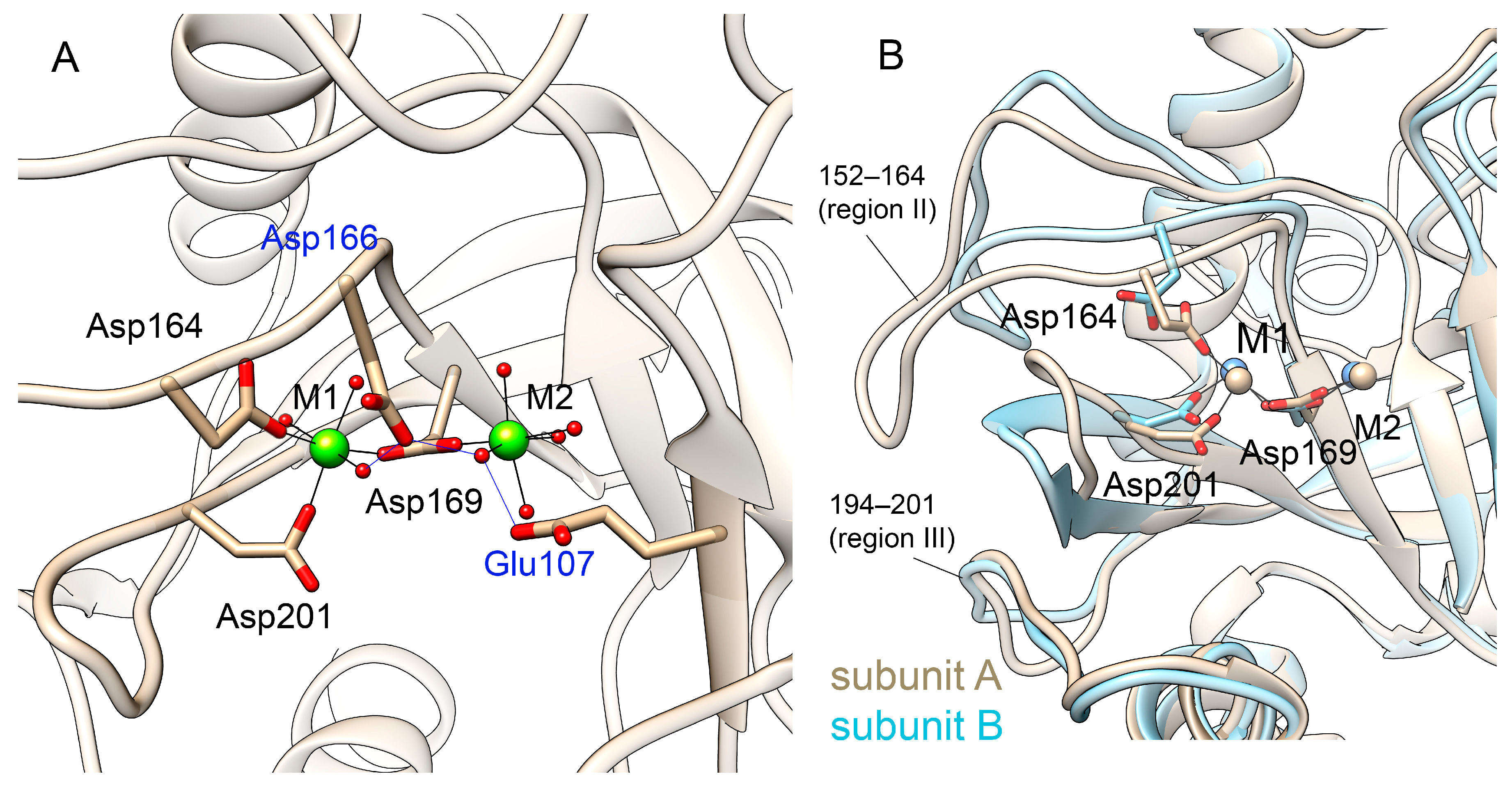
3.2.2. Metal Coordination
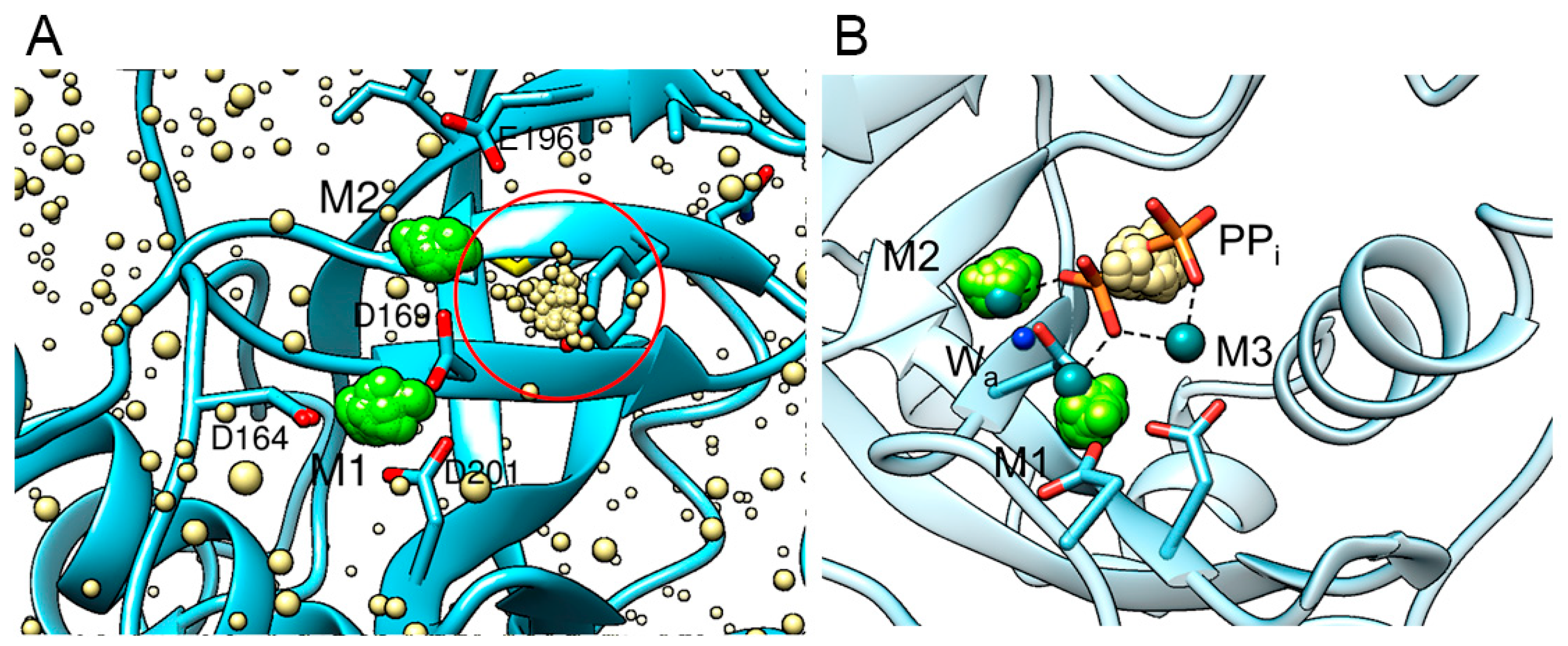
3.2.3. Anionic Binding Site
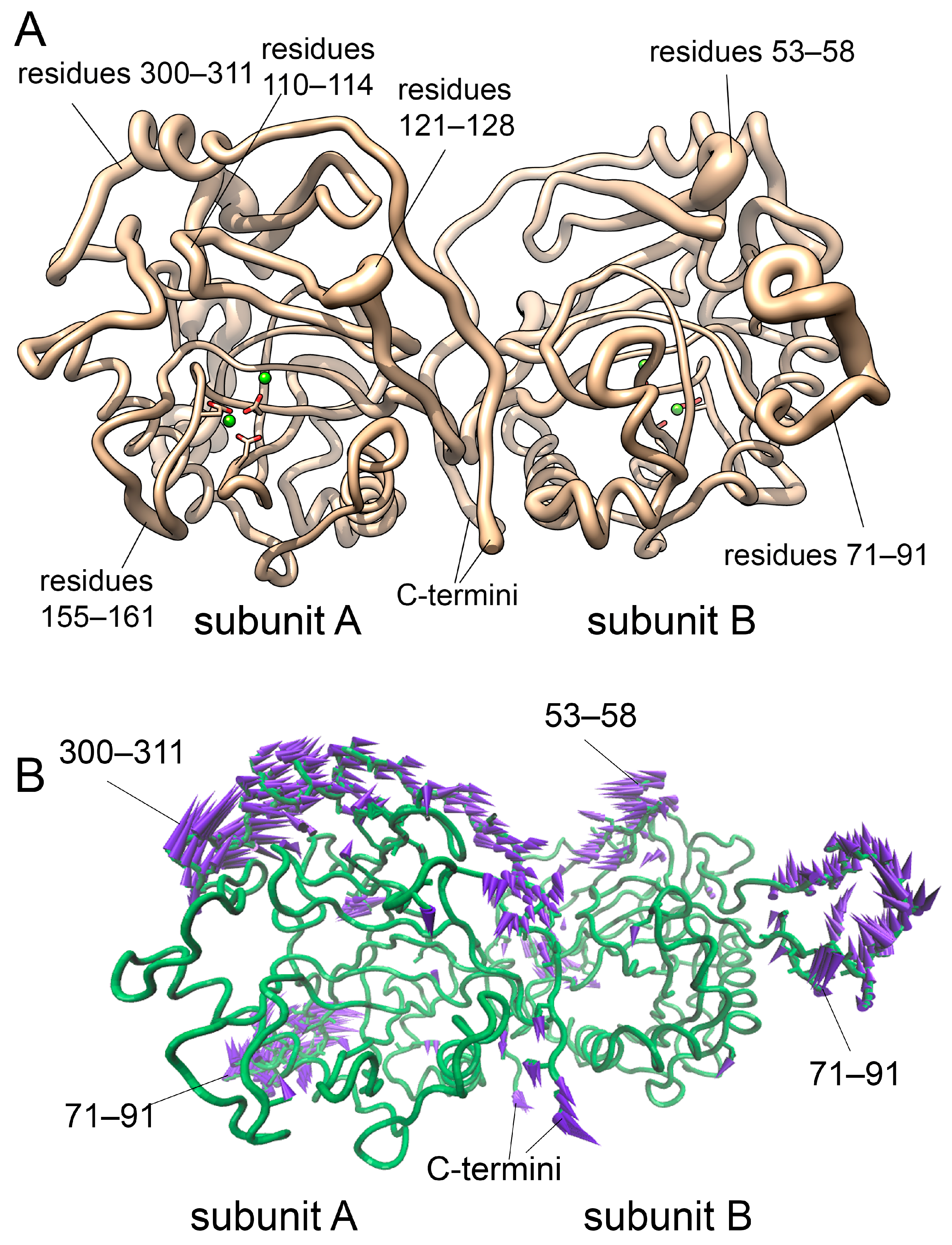
3.2.4. Conformation of the Active Site Loops
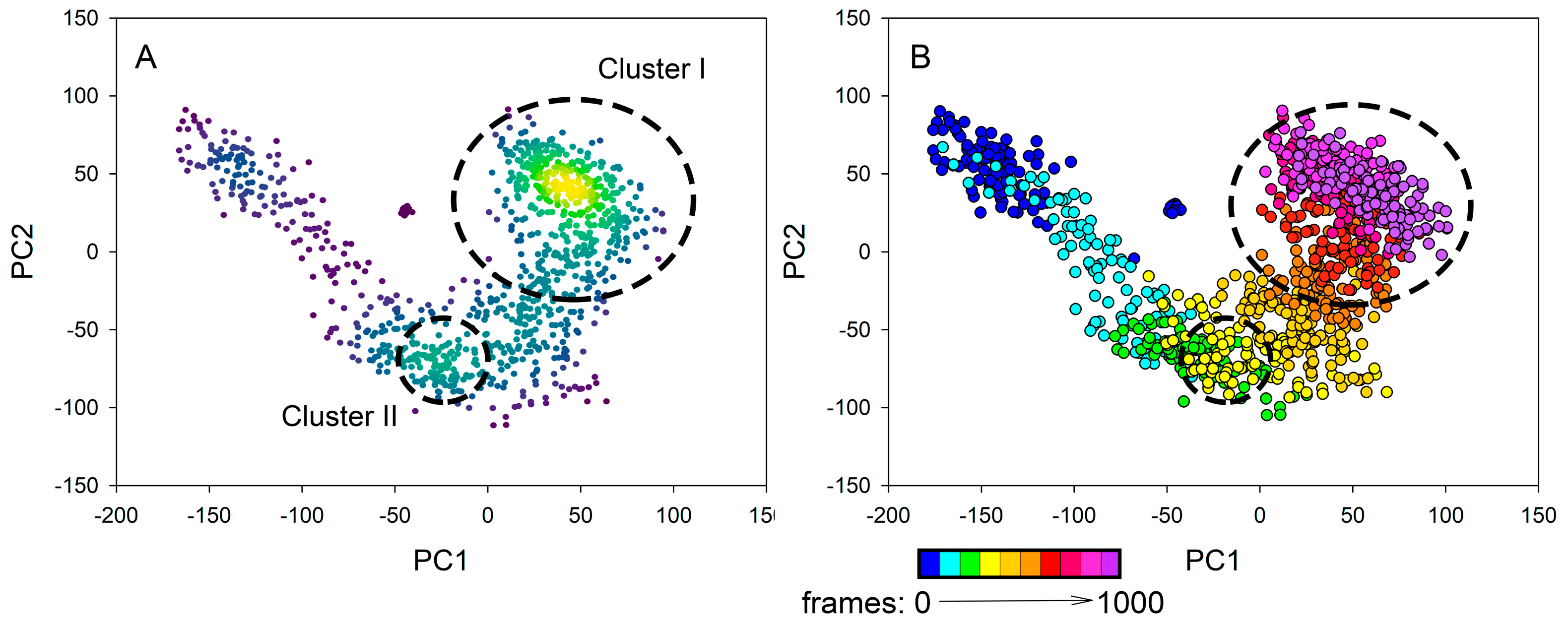
3.2.5. PCA Analysis of MD Trajectory
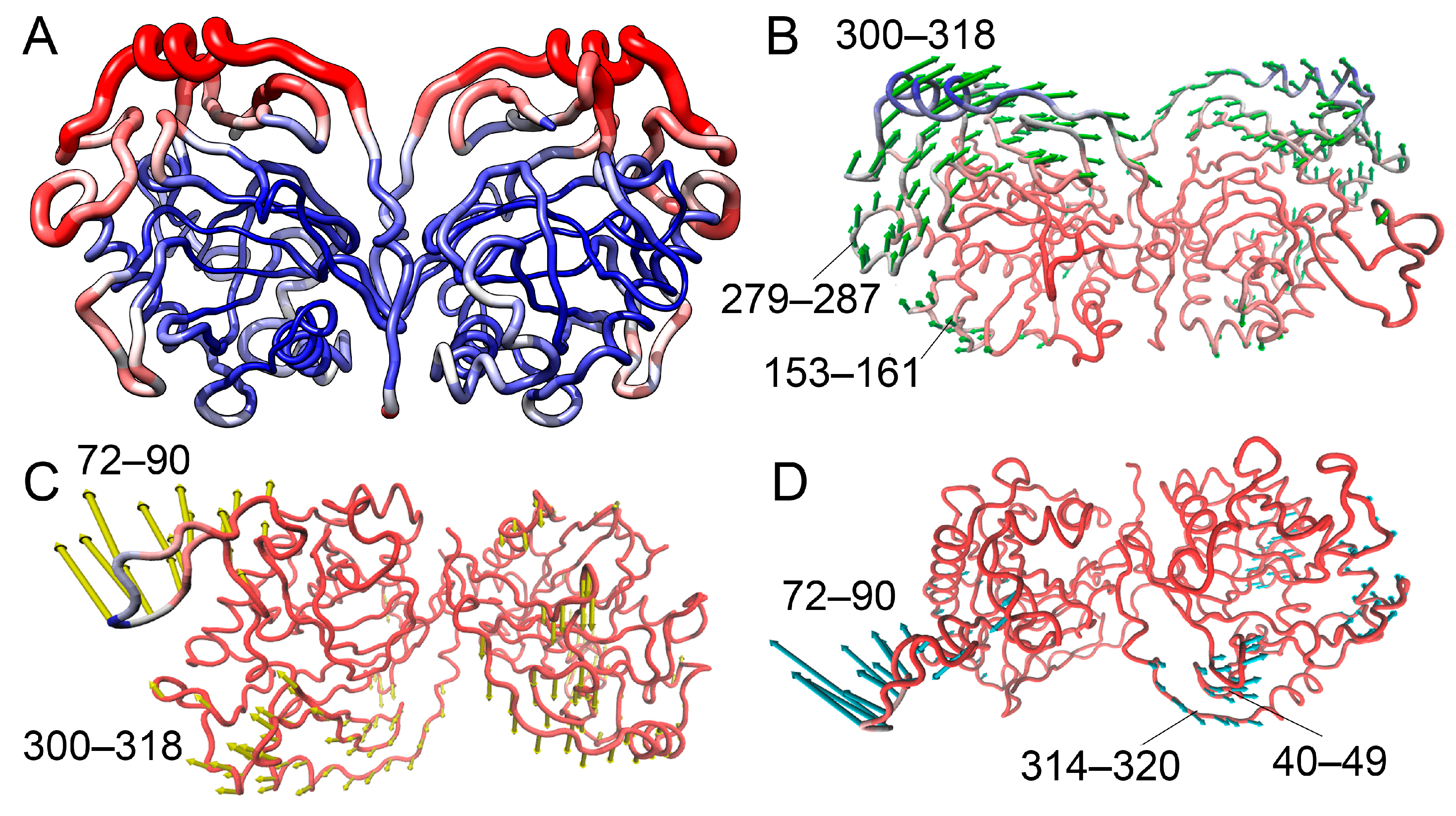
3.2.6. NMA Analysis of hPPA2 Conformational Dynamics
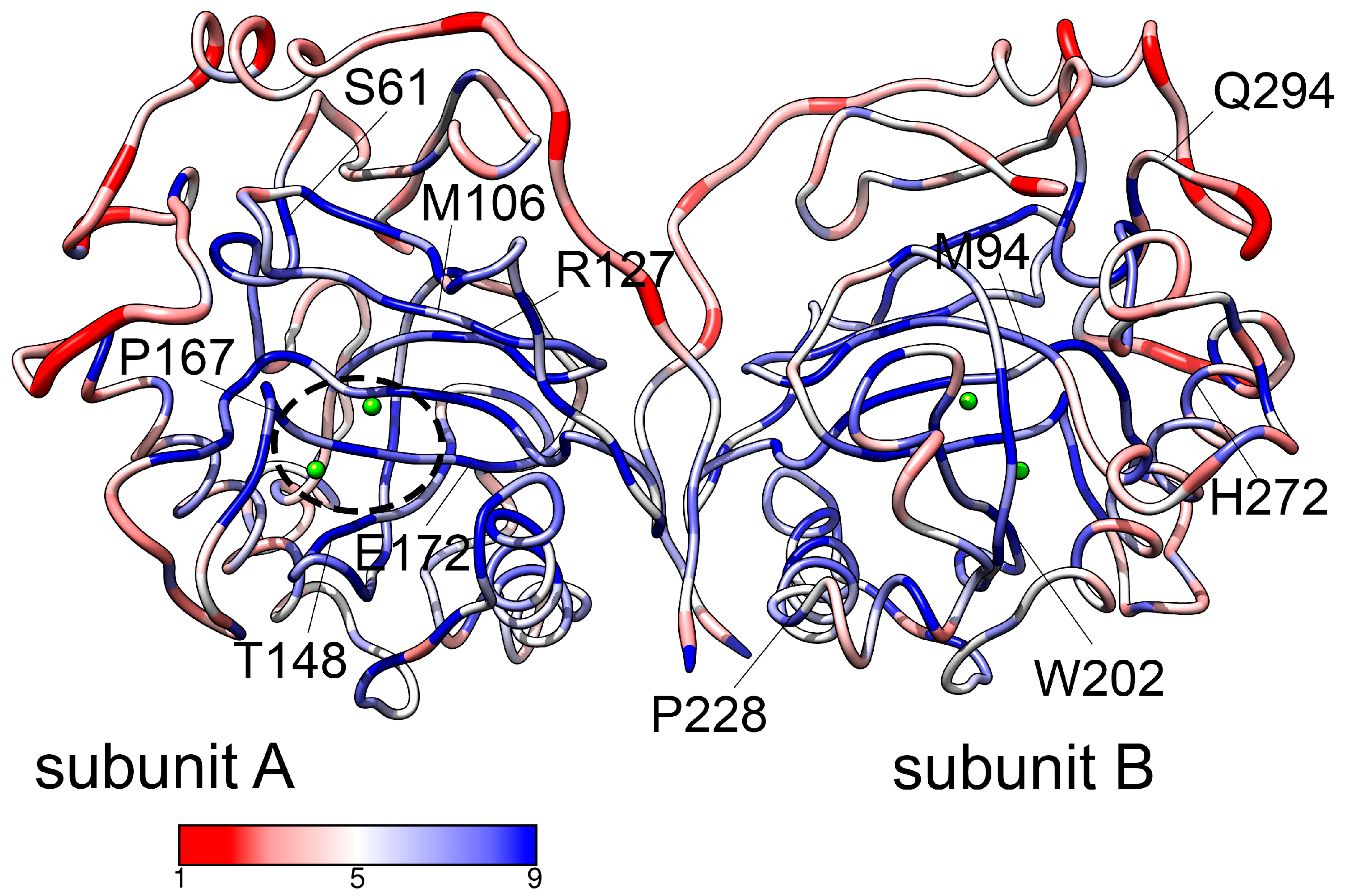
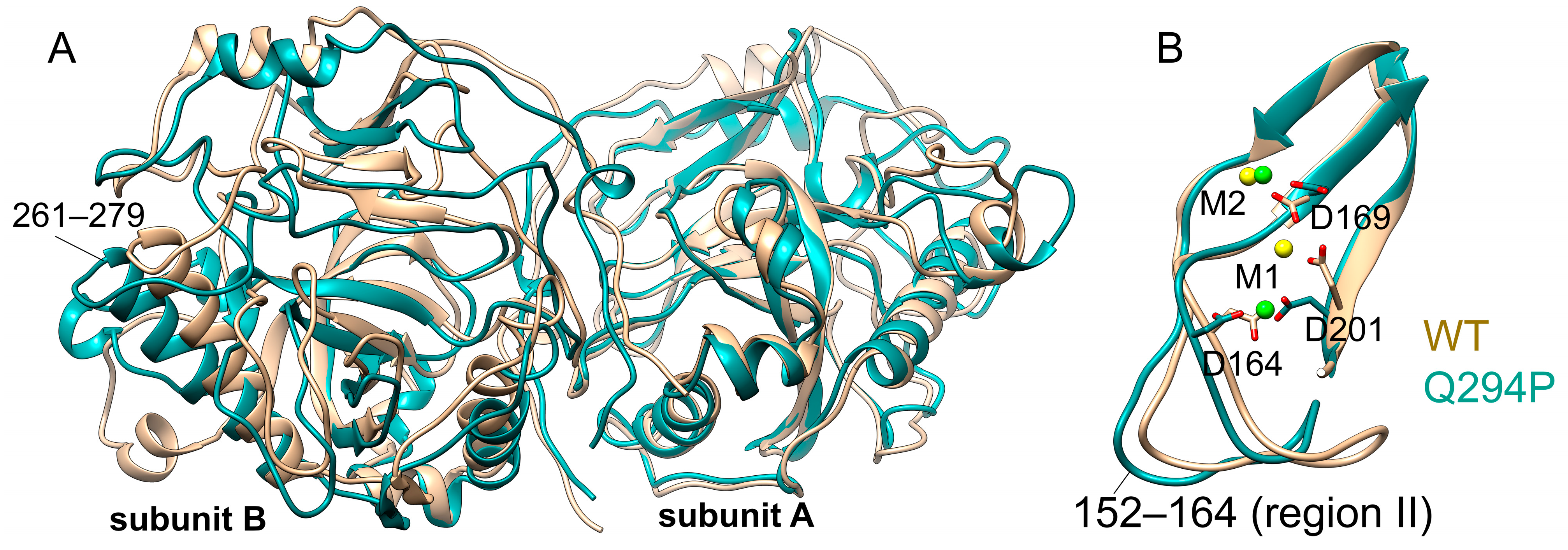
3.3. Effect of Pathogenic Mutations on Conformational Dynamics of hPPA2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Heinonen, J.K. Biological Role of Inorganic Pyrophosphate; Kluwer Academic Publishers Inc.: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Fairchild, T.A.; Patejunas, G. Cloning and expression profile of human inorganic pyrophosphatase. Biochim. Biophys. Acta 1999, 1447, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Curbo, S.; Lagier-Tourenne, C.; Carrozzo, R.; Palenzuela, L.; Lucioli, S.; Zhirano, M.; Santorelli, F.; Arenas, J.; Karlsson, A.; Johansson, M. Human mitochondrial pyrophosphatase: cDNA cloning and analysis of the gene in patients with mt DNA depletion syndromes. Genomics 2006, 87, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Zhu, J.; Dong, L.; Yuan, P.; Zhang, L.; Liu, D. Inorganic pyrophosphatase 1 activates the phosphatidylinositol 3-kinase/Akt signaling to promote tumorigenicity and stemness properties in colorectal cancer. Cell. Signal. 2023, 108, 110693. [Google Scholar] [CrossRef]
- Yang, Y.; Cai, J.; Yin, J.; Wang, D.; Bai, Z.; Zhang, J.; Wang, K.; Yu, G.; Zhang, Z. Inorganic pyrophosphatase (PPA1) is a negative prognostic marker for human gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12482–12490. [Google Scholar]
- Wang, S.; Wei, J.; Li, S.; Luo, Y.; Li, Y.; Wang, X.; Shen, W.; Luo, D.; Liu, D. PPA1, an energy metabolism initiator, plays an important role in the progression of malignant tumors. Front. Oncol. 2022, 12, 1012090. [Google Scholar] [CrossRef]
- Lundin, M.; Baltscheffsky, H.; Ronne, H. Yeast PPA2 gene encodes a mitochondrial inorganic pyrophosphatase that is essential for mitochondrial function. J. Biol. Chem. 1991, 266, 12168–12172. [Google Scholar] [CrossRef]
- Guimier, A.; Gordon, C.T.; Godard, F.; Ravenscroft, G.; Oufadem, M.; Vasnier, C.; Rambaud, C.; Nitschke, P.; Bole-Feysot, C.; Masson, C.; et al. Biallelic PPA2 mutations cause sudden unexpected cardiac arrest in infancy. Am. J. Hum. Genet. 2016, 99, 666–673. [Google Scholar] [CrossRef]
- Kennedy, H.; Haack, T.B.; Hartill, V.; Matakovic, L.; Baumgartner, E.R.; Potter, H.; Mackay, R.; Alston, C.L.; O’Sullivan, S.; McFarland, R.; et al. Sudden cardiac death due to deficiency of the mitochondrial inorganic pyrophosphatase PPA2. Am. J. Hum. Genet. 2016, 99, 674–682. [Google Scholar] [CrossRef]
- Guimier, A.; Achleitner, M.T.; Moreau de Bellaing, A.; Edwards, M.; de Pontual, L.; Mittal, K.; Dunn, K.E.; Grove, M.E.; Tysoe, C.J.; Dimartino, C.; et al. PPA2-associated sudden cardiac death: Extending the clinical and allelic spectrum in 20 new families. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 2415–2425. [Google Scholar] [CrossRef]
- Bezpalaya, E.Y.; Matyuta, I.O.; Vorobyeva, N.N.; Kurilova, S.A.; Oreshkov, S.D.; Minyaev, M.E.; Boyko, K.M.; Rodina, E.V. The crystal structure of yeast mitochondrial type pyrophosphatase provides a model to study pathological mutations in its human ortholog. Biochem. Biophys. Res. Commun. 2024, 738, 150563. [Google Scholar] [CrossRef]
- Oksanen, E.; Ahonen, A.K.; Tuominen, H.; Tuominen, V.; Lahti, R.; Goldman, A.; Heikinheimo, P. A complete structural description of the catalytic cycle of yeast pyrophosphatase. Biochemistry 2007, 46, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Moiseev, V.M.; Rodina, E.V.; Kurilova, S.A.; Vorobyeva, N.N.; Nazarova, T.I.; Avaeva, S.M. Substitutions of glycine residues Gly100 and Gly147 in conservative loops decrease rates of conformational rearrangements of Escherichia coli inorganic pyrophosphatase. Biochemistry 2005, 70, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Samygina, V.R.; Moiseev, V.M.; Rodina, E.V.; Vorobyeva, N.N.; Popov, A.N.; Kurilova, S.A.; Nazarova, T.I.; Avaeva, S.M.; Bartunik, H.D. Reversible Inhibition of Escherichia coli Inorganic Pyrophosphatase by Fluoride: Trapped Catalytic Intermediates in Cryo-crystallographic Studies. J. Mol. Biol. 2007, 366, 1305–1317. [Google Scholar] [CrossRef]
- Baykov, A.A.; Pavlov, A.R.; Kasho, V.N.; Avaeva, S.M. Allosteric regulation of yeast inorganic pyrophosphatase by substrate. Arch. Biochem. Biophys. 1989, 273, 301–308. [Google Scholar] [CrossRef]
- Rodina, E.V.; Vorobyeva, N.N.; Kurilova, S.A.; Sitnik, T.S.; Nazarova, T.I. ATP as effector of inorganic pyrophosphatase of Escherichia coli. The role of residue Lys112 in binding effectors. Biochemistry 2007, 72, 100–108. [Google Scholar] [CrossRef]
- Vorobyeva, N.N.; Kurilova, S.A.; Anashkin, V.A.; Rodina, E.V. Inhibition of Escherichia coli inorganic pyrophosphatase by fructose-1-phosphate. Biochemistry 2017, 82, 953–956. [Google Scholar] [CrossRef]
- Romanov, R.; Kurilova, S.; Baykov, A.; Rodina, E. Effect of structure variations in the inter-subunit contact zone on the activity and allosteric regulation of inorganic pyrophosphatase from Mycobacterium tuberculosis. Biochemistry 2020, 85, 326–333. [Google Scholar] [CrossRef]
- Pang, A.H.; Garzan, A.; Larsen, M.J.; McQuade, T.J.; Garneau-Tsodikova, S.; Tsodikov, O.V. Discovery of Allosteric and Selective Inhibitors of Inorganic Pyrophosphatase from Mycobacterium tuberculosis. ACS Chem. Biol. 2016, 11, 3084–3092. [Google Scholar] [CrossRef]
- Rodina, E.V.; Vorobyeva, N.N.; Kurilova, S.A.; Belenikin, M.S.; Fedorova, N.V.; Nazarova, T.I. ATP as effector of inorganic pyrophosphatase of Escherichia coli. Identification of the binding site for ATP. Biochemistry 2007, 72, 93–99. [Google Scholar] [CrossRef]
- Avaeva, S.; Kurilova, S.; Nazarova, T.; Rodina, E.; Vorobyeva, N.; Sklyankina, V.; Grigorjeva, O.; Harutyunyan, E.; Oganessyan, V.; Wilson, K.; et al. Crystal structure of Escherichia coli inorganic pyrophosphatase complexed with SO42−. Ligand-induced molecular asymmetry. FEBS Lett. 1997, 410, 502–508. [Google Scholar] [CrossRef]
- Quan, L.; Lv, Q.; Zhang, Y. STRUM: Structure-based stability change prediction upon single-point mutation. Bioinformatics 2016, 32, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Yariv, B.; Yariv, E.; Kessel, A.; Masrati, G.; Ben Chorin, A.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Using evolutionary data to make sense of macromolecules with a ’face-lifted’ ConSurf. Protein Sci. 2023, 32, e4582. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Niu, H.; Zhu, J.; Qu, Q.; Zhou, X.; Huang, X.; Du, Z. Crystallographic and modeling study of the human inorganic pyrophosphatase 1: A potential anti-cancer drug target. Proteins 2021, 89, 853–865. [Google Scholar] [CrossRef]
- Heikinheimo, P.; Lehtonen, J.; Baykov, A.; Lahti, R.; Cooperman, B.S.; Goldman, A. The structural basis for pyrophosphatase catalysis. Structure 1996, 4, 1491–1508. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very Fast Empirical Prediction and Rationalization of Protein pKa Values. PROTEINS Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef]
- Eyal, E.; Lum, G.; Bahar, I. The anisotropic network model web server at 2015 (ANM 2.0). Bioinformatics 2015, 31, 1487–1489. [Google Scholar] [CrossRef]
- Yang, L.W.; Rader, A.J.; Liu, X.; Jursa, C.J.; Chen, S.C.; Karimi, H.A.; Bahar, I. o GNM: Online computation of structural dynamics using the Gaussian Network Model. Nucleic Acids Res. 2006, 34, W24–W31. [Google Scholar] [CrossRef]
- Bauer, J.A.; Pavlović, J.; Bauerová-Hlinková, V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules 2019, 24, 3293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | PC1, Å2 | PC2, Å2 |
|---|---|---|
| WT hPPA2 | 4232 | 2431 |
| Ser61Phe | 7709 | 2753 |
| Met94Val | 4419 | 2025 |
| Met106Ile | 7241 | 1738 |
| Gln294Pro | 6163 | 3397 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezpalaya, E.; Kurilova, S.; Vorobyeva, N.; Rodina, E. Conformational Dynamics of Mitochondrial Inorganic Pyrophosphatase hPPA2 and Its Changes Caused by Pathogenic Mutations. Life 2025, 15, 100. https://doi.org/10.3390/life15010100
Bezpalaya E, Kurilova S, Vorobyeva N, Rodina E. Conformational Dynamics of Mitochondrial Inorganic Pyrophosphatase hPPA2 and Its Changes Caused by Pathogenic Mutations. Life. 2025; 15(1):100. https://doi.org/10.3390/life15010100
Chicago/Turabian StyleBezpalaya, Ekaterina, Svetlana Kurilova, Nataliya Vorobyeva, and Elena Rodina. 2025. "Conformational Dynamics of Mitochondrial Inorganic Pyrophosphatase hPPA2 and Its Changes Caused by Pathogenic Mutations" Life 15, no. 1: 100. https://doi.org/10.3390/life15010100
APA StyleBezpalaya, E., Kurilova, S., Vorobyeva, N., & Rodina, E. (2025). Conformational Dynamics of Mitochondrial Inorganic Pyrophosphatase hPPA2 and Its Changes Caused by Pathogenic Mutations. Life, 15(1), 100. https://doi.org/10.3390/life15010100