
Advances and Mechanisms of RNA–Ligand Interaction Predictions


Abstract
:1. Introduction
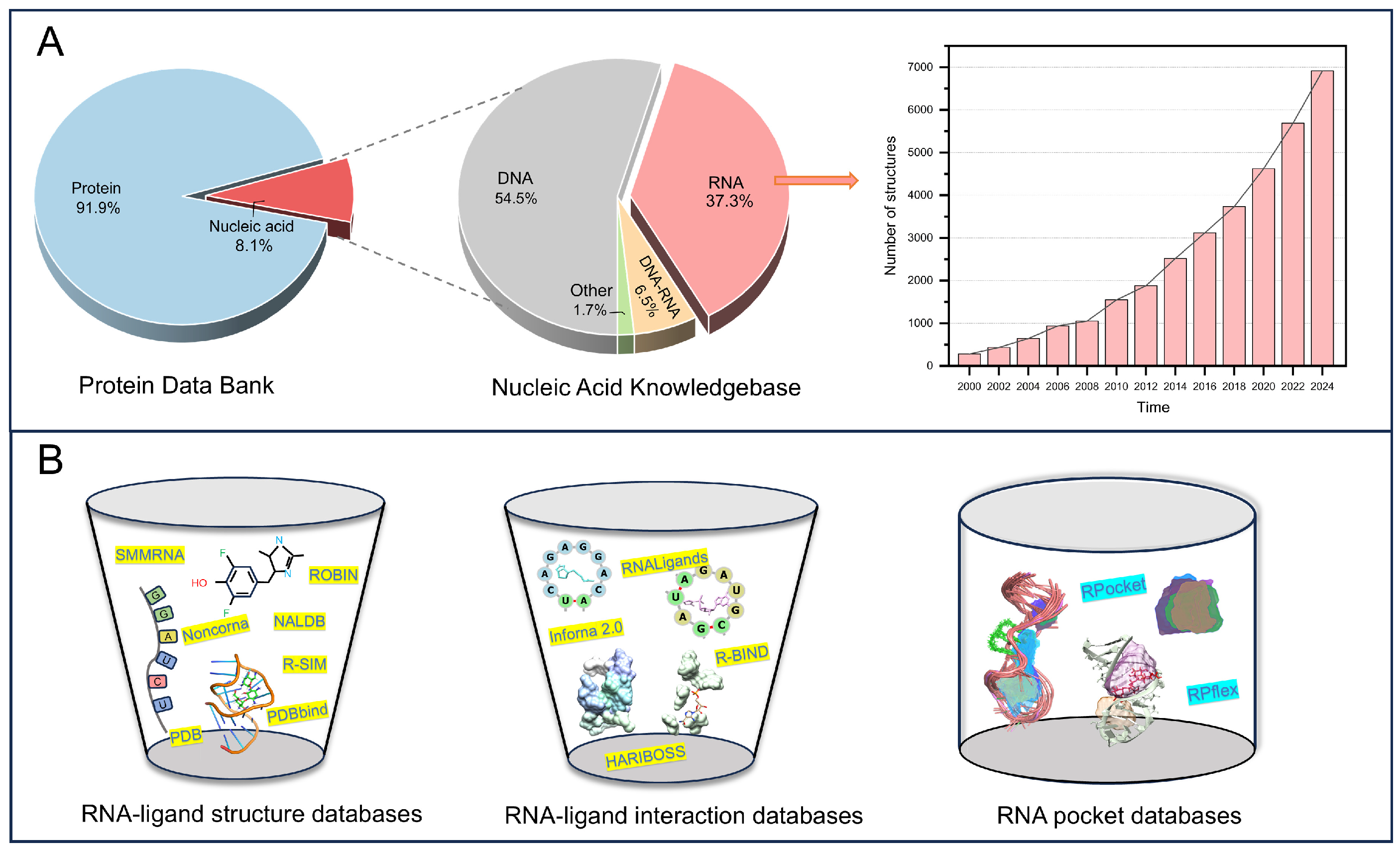
2. RNA–Ligand Database Resources
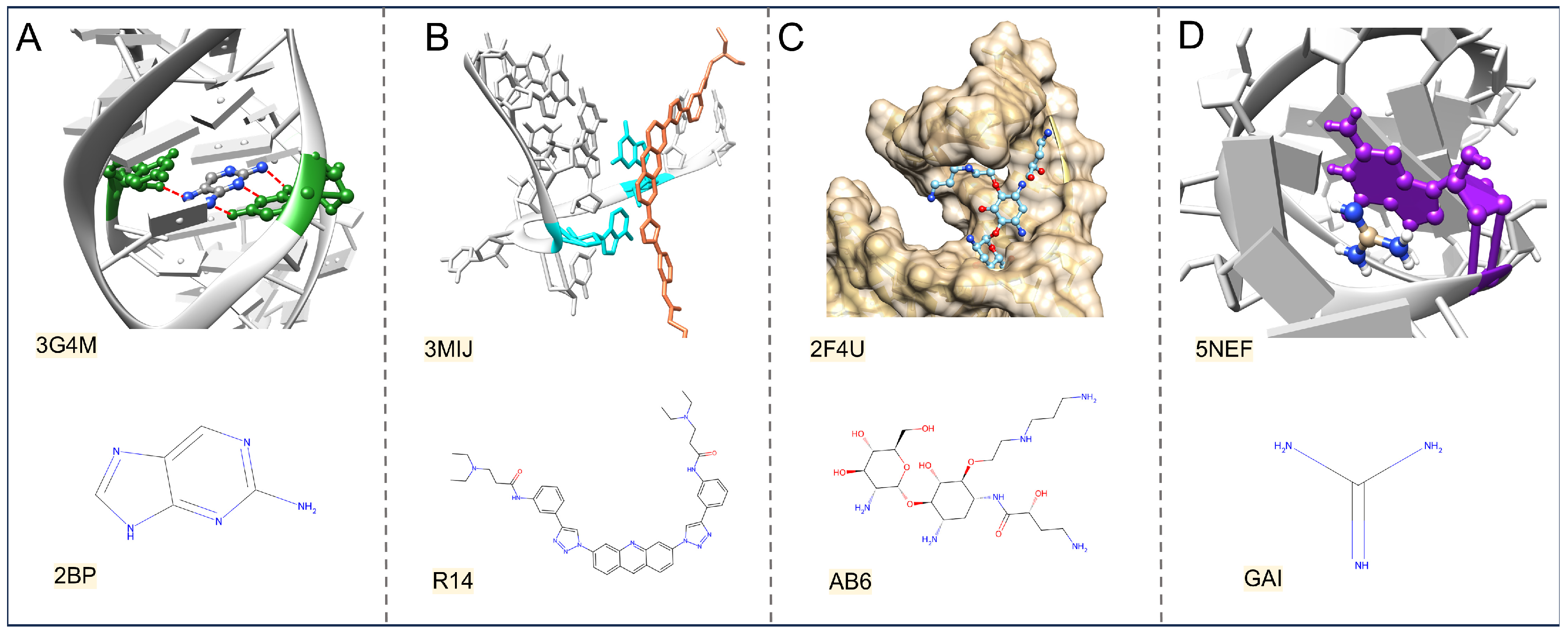
3. Physics-Based Interaction Forces on RNA–Ligand Complexes
3.1. The Short-Range Forces
3.2. The Long-Range Force
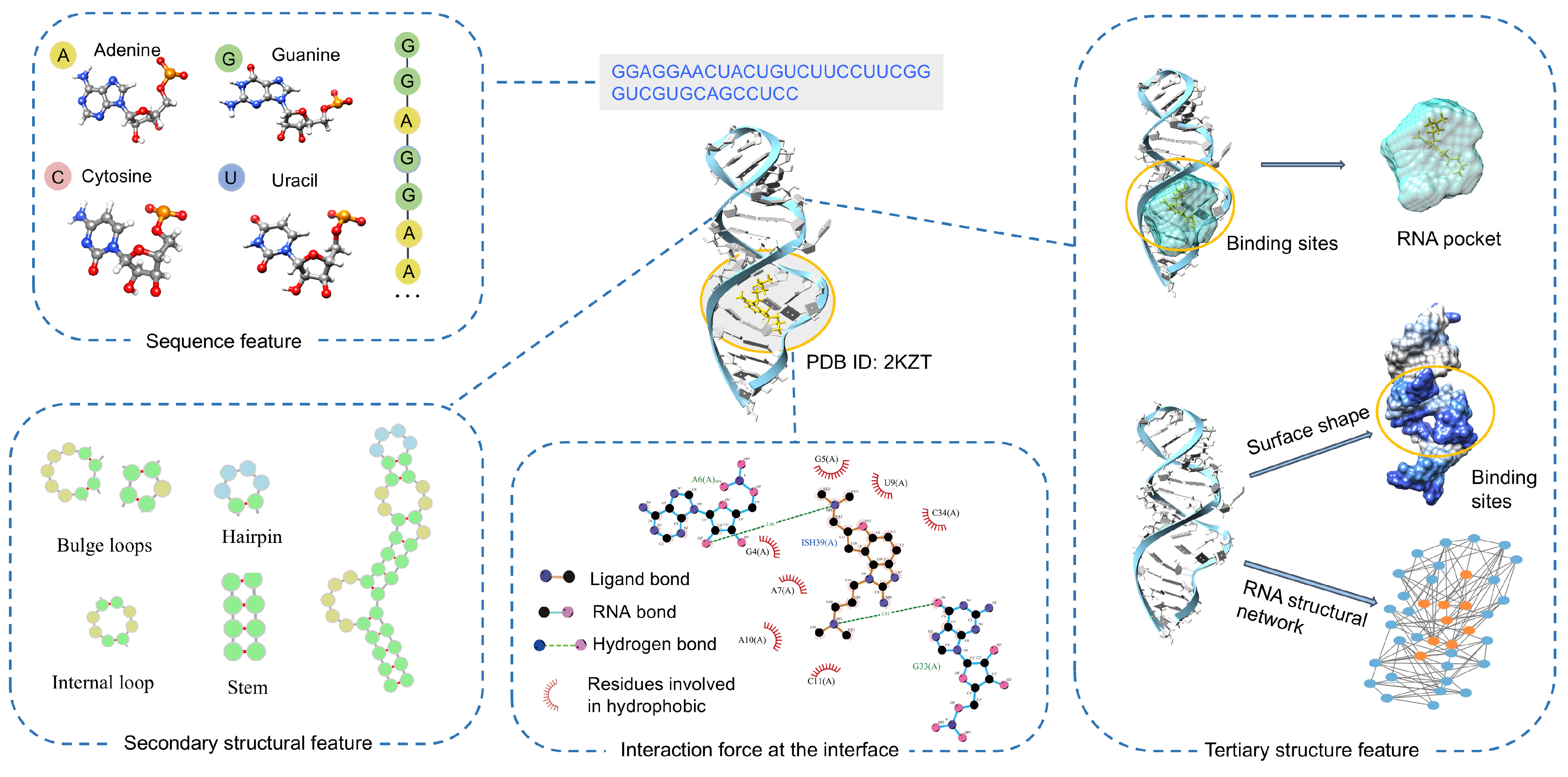
4. Interaction Mechanisms Extracted from RNA Features
4.1. Sequence Features
4.2. Secondary Structure Features
4.3. Tertiary Structure Features
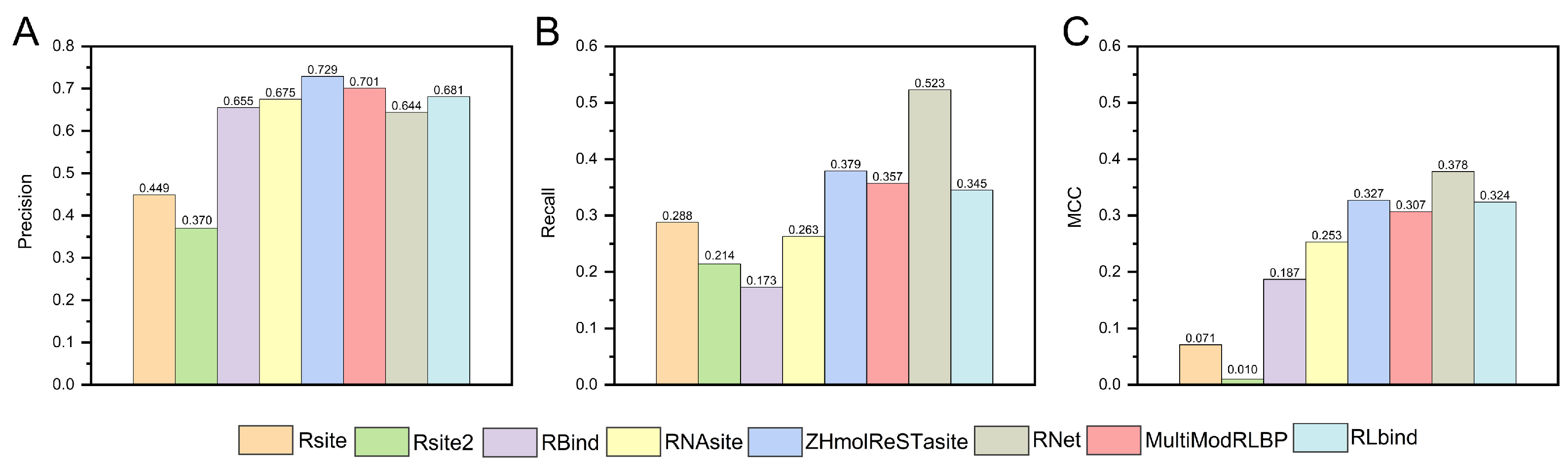
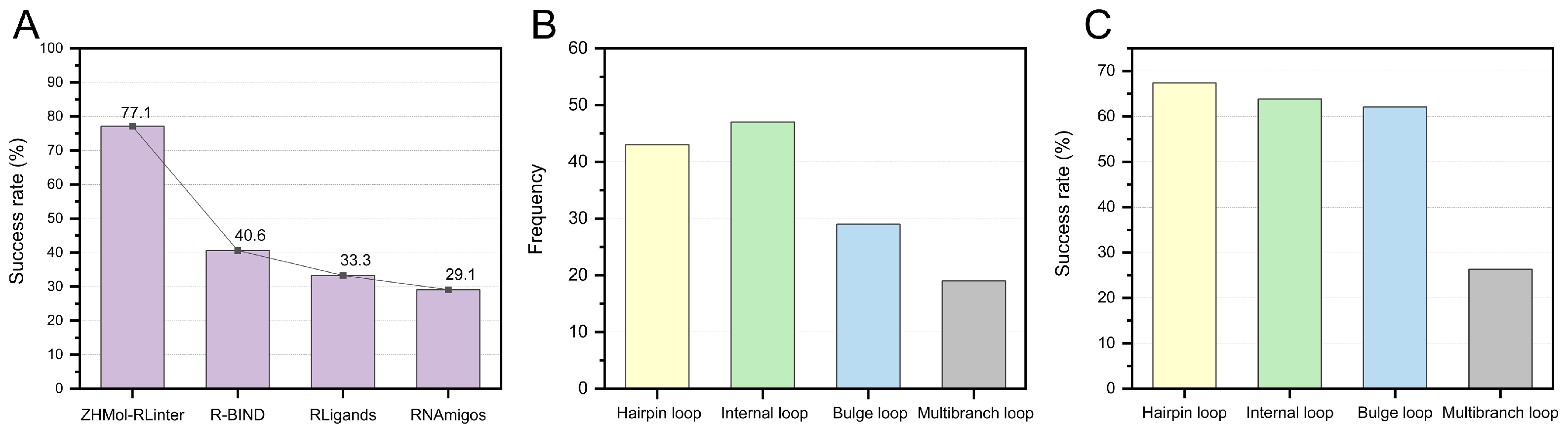
5. RNA–Ligand Interaction Prediction
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Serganov, A.; Huang, L.; Patel, D.J. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature 2009, 458, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Loan Young, T.; Chang Wang, K.; James Varley, A.; Li, B. Clinical delivery of circular RNA: Lessons learned from RNA drug development. Adv. Drug Deliv. Rev. 2023, 197, 114826. [Google Scholar] [CrossRef]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, A.E. Small molecule-RNA targeting: Starting with the fundamentals. Chem. Commun. 2020, 56, 14744–14756. [Google Scholar] [CrossRef]
- Dibrov, S.M.; Ding, K.; Brunn, N.D.; Parker, M.A.; Bergdahl, B.M.; Wyles, D.L.; Hermann, T. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc. Natl. Acad. Sci. USA 2012, 109, 5223–5228. [Google Scholar] [CrossRef]
- Andreasson, J.O.L.; Savinov, A.; Block, S.M.; Greenleaf, W.J. Comprehensive sequence-to-function mapping of cofactor-dependent RNA catalysis in the glmS ribozyme. Nat. Commun. 2020, 11, 1663. [Google Scholar] [CrossRef]
- Bosshard, H.R. Molecular recognition by induced fit: How fit is the concept? Physiology 2001, 16, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Duchardt-Ferner, E.; Weigand, J.E.; Ohlenschlager, O.; Schmidtke, S.R.; Suess, B.; Wohnert, J. Highly modular structure and ligand binding by conformational capture in a minimalistic riboswitch. Angew. Chem. Int. Ed. Engl. 2010, 49, 6216–6219. [Google Scholar] [CrossRef]
- Leulliot, N.; Varani, G. Current topics in RNA-protein recognition: Control of specificity and biological function through induced fit and conformational capture. Biochemistry 2001, 40, 7947–7956. [Google Scholar] [CrossRef] [PubMed]
- Haller, A.; Souliere, M.F.; Micura, R. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc. Chem. Res. 2011, 44, 1339–1348. [Google Scholar] [CrossRef]
- Haller, A.; Rieder, U.; Aigner, M.; Blanchard, S.C.; Micura, R. Conformational capture of the SAM-II riboswitch. Nat. Chem. Biol. 2011, 7, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Fernandez, X.M. The 2024 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 2024, 52, D1–D9. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Lawson, C.L.; Schneider, B. Developing Community Resources for Nucleic Acid Structures. Life 2022, 12, 540. [Google Scholar] [CrossRef]
- Mehta, A.; Sonam, S.; Gouri, I.; Loharch, S.; Sharma, D.K.; Parkesh, R. SMMRNA: A database of small molecule modulators of RNA. Nucleic Acids Res. 2014, 42, D132–D141. [Google Scholar] [CrossRef]
- Kumar Mishra, S.; Kumar, A. NALDB: Nucleic acid ligand database for small molecules targeting nucleic acid. Database 2016, 2016, baw002. [Google Scholar] [CrossRef]
- Zhang, W.; Yao, G.; Wang, J.; Yang, M.; Wang, J.; Zhang, H.; Li, W. ncRPheno: A comprehensive database platform for identification and validation of disease related noncoding RNAs. RNA Biol. 2020, 17, 943–955. [Google Scholar] [CrossRef]
- Cheng, J.; Lin, Y.; Xu, L.; Chen, K.; Li, Q.; Xu, K.; Ning, L.; Kang, J.; Cui, T.; Huang, Y.; et al. ViRBase v3.0: A virus and host ncRNA-associated interaction repository with increased coverage and annotation. Nucleic Acids Res. 2022, 50, D928–D933. [Google Scholar] [CrossRef]
- Lin, X.; Lu, Y.; Zhang, C.; Cui, Q.; Tang, Y.D.; Ji, X.; Cui, C. LncRNADisease v3.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2024, 52, D1365–D1369. [Google Scholar] [CrossRef]
- Szpotkowski, K.; Wojcik, K.; Kurzynska-Kokorniak, A. Structural studies of protein-nucleic acid complexes: A brief overview of the selected techniques. Comput. Struct. Biotechnol. J. 2023, 21, 2858–2872. [Google Scholar] [CrossRef]
- Schmidt, M. Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. Int. J. Mol. Sci. 2019, 20, 1401. [Google Scholar] [CrossRef]
- Danev, R.; Yanagisawa, H.; Kikkawa, M. Cryo-Electron Microscopy Methodology: Current Aspects and Future Directions. Trends Biochem. Sci. 2019, 44, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gong, Z.; Zhao, Y. Methods and Applications in Proteins and RNAs. Life 2023, 13, 672. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The Protein Data Bank Archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.E.; Zou, J.Y.; Chang, H. Machine learning modeling of RNA structures: Methods, challenges and future perspectives. Brief. Bioinform. 2023, 24, bbad210. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Yang, X.; Zhang, Y.; Zhang, Z.; Zhang, G.; Zhang, B.T. RNA-targeted small-molecule drug discoveries: A machine-learning perspective. RNA Biol. 2023, 20, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhuo, C.; Gao, J.; Zeng, C.; Zhao, Y. AI-integrated network for RNA complex structure and dynamic prediction. Biophys. Rev. 2024, 5, 041304. [Google Scholar] [CrossRef]
- Justyna, M.; Antczak, M.; Szachniuk, M. Machine learning for RNA 2D structure prediction benchmarked on experimental data. Brief. Bioinform. 2023, 24, bbad153. [Google Scholar] [CrossRef]
- Schneider, B.; Sweeney, B.A.; Bateman, A.; Cerny, J.; Zok, T.; Szachniuk, M. When will RNA get its AlphaFold moment? Nucleic Acids Res. 2023, 51, 9522–9532. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Postic, G.; Ghannay, S.; Tahi, F. State-of-the-RNArt: Benchmarking current methods for RNA 3D structure prediction. NAR Genom. Bioinform. 2024, 6, lqae048. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jian, Y.; Hou, J.; Zeng, C.; Zhao, Y. RNet: A network strategy to predict RNA binding preferences. Brief. Bioinform. 2023, 25, bbad482. [Google Scholar] [CrossRef]
- Su, H.; Peng, Z.; Yang, J. Recognition of small molecule-RNA binding sites using RNA sequence and structure. Bioinformatics 2021, 37, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Jian, Y.; Vosoughi, S.; Zeng, C.; Zhao, Y. Evaluating native-like structures of RNA-protein complexes through the deep learning method. Nat. Commun. 2023, 14, 1060. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhuo, C.; Gao, J.; Liu, H.; Zhao, Y. Advances and Challenges in Scoring Functions for RNA-Protein Complex Structure Prediction. Biomolecules 2024, 14, 1245. [Google Scholar] [CrossRef]
- Gao, J.; Liu, H.; Zhuo, C.; Zeng, C.; Zhao, Y. Predicting Small Molecule Binding Nucleotides in RNA Structures Using RNA Surface Topography. J. Chem. Inf. Model. 2024, 64, 6979–6992. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Quan, L.; Jin, Z.; Wu, H.; Ma, X.; Wang, X.; Xie, J.; Pan, D.; Chen, T.; Wu, T.; et al. MultiModRLBP: A Deep Learning Approach for Multi-Modal RNA-Small Molecule Ligand Binding Sites Prediction. IEEE J. Biomed. Health Inform. 2024, 28, 4995–5006. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gao, L. Contrastive pre-training and 3D convolution neural network for RNA and small molecule binding affinity prediction. Bioinformatics 2024, 40, btae155. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Roy, A.; Gromiha, M.M. Reliable method for predicting the binding affinity of RNA-small molecule interactions using machine learning. Brief. Bioinform. 2024, 25, bbae002. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.L.; Berman, H.M.; Chen, L.; Vallat, B.; Zirbel, C.L. The Nucleic Acid Knowledgebase: A new portal for 3D structural information about nucleic acids. Nucleic Acids Res. 2024, 52, D245–D254. [Google Scholar] [CrossRef]
- Li, L.; Wu, P.; Wang, Z.; Meng, X.; Zha, C.; Li, Z.; Qi, T.; Zhang, Y.; Han, B.; Li, S.; et al. NoncoRNA: A database of experimentally supported non-coding RNAs and drug targets in cancer. J. Hematol. Oncol. 2020, 13, 15. [Google Scholar] [CrossRef]
- Yazdani, K.; Jordan, D.; Yang, M.; Fullenkamp, C.R.; Calabrese, D.R.; Boer, R.; Hilimire, T.; Allen, T.E.H.; Khan, R.T.; Schneekloth, J.S., Jr. Machine Learning Informs RNA-Binding Chemical Space. Angew. Chem. Int. Ed. Engl. 2023, 62, e202211358. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [CrossRef]
- Ramaswamy Krishnan, S.; Roy, A.; Michael Gromiha, M. R-SIM: A Database of Binding Affinities for RNA-small Molecule Interactions. J. Mol. Biol. 2023, 435, 167914. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, J.; Zhang, Z. RNALigands: A database and web server for RNA-ligand interactions. RNA 2022, 28, 115–122. [Google Scholar] [CrossRef]
- Disney, M.D.; Winkelsas, A.M.; Velagapudi, S.P.; Southern, M.; Fallahi, M.; Childs-Disney, J.L. Inforna 2.0: A Platform for the Sequence-Based Design of Small Molecules Targeting Structured RNAs. ACS Chem. Biol. 2016, 11, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Donlic, A.; Swanson, E.G.; Chiu, L.Y.; Wicks, S.L.; Juru, A.U.; Cai, Z.; Kassam, K.; Laudeman, C.; Sanaba, B.G.; Sugarman, A.; et al. R-BIND 2.0: An Updated Database of Bioactive RNA-Targeting Small Molecules and Associated RNA Secondary Structures. ACS Chem. Biol. 2022, 17, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Panei, F.P.; Torchet, R.; Menager, H.; Gkeka, P.; Bonomi, M. HARIBOSS: A curated database of RNA-small molecules structures to aid rational drug design. Bioinformatics 2022, 38, 4185–4193. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, H.; Zeng, C.; Zhao, Y. RPocket: An intuitive database of RNA pocket topology information with RNA-ligand data resources. BMC Bioinform. 2021, 22, 428. [Google Scholar] [CrossRef]
- Haque, F.; Pi, F.; Zhao, Z.; Gu, S.; Hu, H.; Yu, H.; Guo, P. RNA versatility, flexibility, and thermostability for practice in RNA nanotechnology and biomedical applications. Wiley Interdiscip. Rev. RNA 2018, 9, e1452. [Google Scholar] [CrossRef]
- Zhuo, C.; Zeng, C.; Yang, R.; Liu, H.; Zhao, Y. RPflex: A Coarse-Grained Network Model for RNA Pocket Flexibility Study. Int. J. Mol. Sci. 2023, 24, 5497. [Google Scholar] [CrossRef]
- Auweter, S.D.; Oberstrass, F.C.; Allain, F.H. Sequence-specific binding of single-stranded RNA: Is there a code for recognition? Nucleic Acids Res. 2006, 34, 4943–4959. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Schwerdtfeger, P.; Wales, D.J. 100 Years of the Lennard-Jones Potential. J. Chem. Theory Comput. 2024, 20, 3379–3405. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.D.; Reyes, F.E.; Edwards, A.L.; Batey, R.T. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 2009, 17, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Seelam, P.P.; Mitra, A.; Sharma, P. Pairing interactions between nucleobases and ligands in aptamer:ligand complexes of riboswitches: Crystal structure analysis, classification, optimal structures, and accurate interaction energies. RNA 2019, 25, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Padroni, G.; Patwardhan, N.N.; Schapira, M.; Hargrove, A.E. Systematic analysis of the interactions driving small molecule-RNA recognition. RSC Med. Chem. 2020, 11, 802–813. [Google Scholar] [CrossRef]
- Wilson, K.A.; Holland, D.J.; Wetmore, S.D. Topology of RNA-protein nucleobase-amino acid pi-pi interactions and comparison to analogous DNA-protein pi-pi contacts. RNA 2016, 22, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Sparapani, S.; Parkinson, G.N.; Neidle, S. Structural basis of telomeric RNA quadruplex—Acridine ligand recognition. J. Am. Chem. Soc. 2011, 133, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Onofrio, A.; Parisi, G.; Punzi, G.; Todisco, S.; Di Noia, M.A.; Bossis, F.; Turi, A.; De Grassi, A.; Pierri, C.L. Distance-dependent hydrophobic-hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys. 2014, 16, 18907–18917. [Google Scholar] [CrossRef]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The protein folding problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef]
- Nandy, A.; Shekhar, S.; Paul, B.K.; Mukherjee, S. Exploring the Nucleobase-Specific Hydrophobic Interaction of Cryptolepine Hydrate with RNA and Its Subsequent Sequestration. Langmuir 2021, 37, 11176–11187. [Google Scholar] [CrossRef]
- Dezanet, C.; Kempf, J.; Mingeot-Leclercq, M.P.; Decout, J.L. Amphiphilic Aminoglycosides as Medicinal Agents. Int. J. Mol. Sci. 2020, 21, 7411. [Google Scholar] [CrossRef]
- Murray, J.B.; Meroueh, S.O.; Russell, R.J.; Lentzen, G.; Haddad, J.; Mobashery, S. Interactions of designer antibiotics and the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2006, 13, 129–138. [Google Scholar] [CrossRef]
- Koehl, P. Electrostatics calculations: Latest methodological advances. Curr. Opin. Struct. Biol. 2006, 16, 142–151. [Google Scholar] [CrossRef]
- Jin, S.; Wang, D.; Jin, X.; Chen, G.Z. Intramolecular electrostatics: Coulomb’s law at sub-nanometers. Chemphyschem 2004, 5, 1623–1629. [Google Scholar] [CrossRef]
- Rooman, M.; Lievin, J.; Buisine, E.; Wintjens, R. Cation-pi/H-bond stair motifs at protein-DNA interfaces. J. Mol. Biol. 2002, 319, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, J.; Lilley, D.M.J. The Structure of the Guanidine-II Riboswitch. Cell Chem. Biol. 2017, 24, 695–702.E2. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cao, Z.; Chen, C.; Li, H.; He, S.; Hou, X.; Liang, M.; Yang, X.; Wang, J. Nanoassembly of doxorubicin-conjugated polyphosphoester and siRNA simultaneously elicited macrophage- and T cell-mediated anticancer immune response for cancer therapy. Biomaterials 2023, 302, 122339. [Google Scholar] [CrossRef]
- Krepl, M.; Vogele, J.; Kruse, H.; Duchardt-Ferner, E.; Wohnert, J.; Sponer, J. An intricate balance of hydrogen bonding, ion atmosphere and dynamics facilitates a seamless uracil to cytosine substitution in the U-turn of the neomycin-sensing riboswitch. Nucleic Acids Res. 2018, 46, 6528–6543. [Google Scholar] [CrossRef]
- Shih, P.; Pedersen, L.G.; Gibbs, P.R.; Wolfenden, R. Hydrophobicities of the nucleic acid bases: Distribution coefficients from water to cyclohexane. J. Mol. Biol. 1998, 280, 421–430. [Google Scholar] [CrossRef]
- Roth, A.; Breaker, R.R. The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 2009, 78, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yildirim, I.; Park, H.; Lohman, J.R.; Guan, L.; Tran, T.; Sarkar, P.; Schatz, G.C.; Disney, M.D. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem. Biol. 2014, 9, 538–550. [Google Scholar] [CrossRef]
- Liu, B.; Childs-Disney, J.L.; Znosko, B.M.; Wang, D.; Fallahi, M.; Gallo, S.M.; Disney, M.D. Analysis of secondary structural elements in human microRNA hairpin precursors. BMC Bioinform. 2016, 17, 112. [Google Scholar] [CrossRef]
- Wang, K.; Jian, Y.; Wang, H.; Zeng, C.; Zhao, Y. RBind: Computational network method to predict RNA binding sites. Bioinformatics 2018, 34, 3131–3136. [Google Scholar] [CrossRef]
- Voss, N.R.; Gerstein, M. 3V: Cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010, 38, W555–W562. [Google Scholar] [CrossRef]
- Kawabata, T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins 2010, 78, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.V.S.; Ribeiro-Filho, H.V.; Pereira, J.G.C.; Lopes-de-Oliveira, P.S. KVFinder-web: A web-based application for detecting and characterizing biomolecular cavities. Nucleic Acids Res. 2023, 51, W289–W297. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; C, N.; Pilla, S.P.; Bahadur, R.P. Molecular architecture of protein-RNA recognition sites. J. Biomol. Struct. Dyn. 2015, 33, 2738–2751. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, R.P.; Zacharias, M.; Janin, J. Dissecting protein-RNA recognition sites. Nucleic Acids Res. 2008, 36, 2705–2716. [Google Scholar] [CrossRef]
- Sato, K.; Hamada, M. Recent trends in RNA informatics: A review of machine learning and deep learning for RNA secondary structure prediction and RNA drug discovery. Brief. Bioinform. 2023, 24, bbad186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, S.J. Advances in machine-learning approaches to RNA-targeted drug design. Artif. Intell. Chem. 2024, 2, 100053. [Google Scholar] [CrossRef]
- Morishita, E.C.; Nakamura, S. Recent applications of artificial intelligence in RNA-targeted small molecule drug discovery. Expert Opin. Drug Discov. 2024, 19, 415–431. [Google Scholar] [CrossRef]
- Zeng, P.; Li, J.; Ma, W.; Cui, Q. Rsite: A computational method to identify the functional sites of noncoding RNAs. Sci. Rep. 2015, 5, 9179. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Cui, Q. Rsite2: An efficient computational method to predict the functional sites of noncoding RNAs. Sci. Rep. 2016, 6, 19016. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, R.; Wu, Y.; Li, M. RLBind: A deep learning method to predict RNA-ligand binding sites. Brief. Bioinform. 2023, 24, bbac486. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.; Mallet, V.; Gendron, R.S.; Reinharz, V.; Hamilton, W.L.; Moitessier, N.; Waldispuhl, J. Augmented base pairing networks encode RNA-small molecule binding preferences. Nucleic Acids Res. 2020, 48, 7690–7699. [Google Scholar] [CrossRef]
- Zhuo, C.; Gao, J.; Li, A.; Liu, X.; Zhao, Y. A Machine Learning Method for RNA-Small Molecule Binding Preference Prediction. J. Chem. Inf. Model. 2024, 64, 7386–7397. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Studer, G.; Robin, X.; Bienert, S.; Tauriello, G.; Schwede, T. The structure assessment web server: For proteins, complexes and more. Nucleic Acids Res. 2024, 52, W318–W323. [Google Scholar] [CrossRef]
- Lukasiak, P.; Antczak, M.; Ratajczak, T.; Szachniuk, M.; Popenda, M.; Adamiak, R.W.; Blazewicz, J. RNAssess—A web server for quality assessment of RNA 3D structures. Nucleic Acids Res. 2015, 43, W502–W506. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, C.; Wallner, B. DockQ v2: Improved automatic quality measure for protein multimers, nucleic acids, and small molecules. Bioinformatics 2024, 40, btae586. [Google Scholar] [CrossRef]
- Nithin, C.; Kmiecik, S.; Blaszczyk, R.; Nowicka, J.; Tuszynska, I. Comparative analysis of RNA 3D structure prediction methods: Towards enhanced modeling of RNA-ligand interactions. Nucleic Acids Res. 2024, 52, 7465–7486. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Das, R.; Millett, I.S.; Herschlag, D.; Doniach, S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc. Natl. Acad. Sci. USA 2005, 102, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.B.; Herschlag, D. Unwinding RNA’s secrets: Advances in the biology, physics, and modeling of complex RNAs. Curr. Opin. Struct. Biol. 2008, 18, 305–314. [Google Scholar] [CrossRef]
- Sun, L.Z.; Zhang, J.X.; Chen, S.J. MCTBI: A web server for predicting metal ion effects in RNA structures. RNA 2017, 23, 1155–1165. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, D.; Jiang, Y.; Chen, S.J. Modeling Loop Composition and Ion Concentration Effects in RNA Hairpin Folding Stability. Biophys. J. 2020, 119, 1439–1455. [Google Scholar] [CrossRef]
- Xu, J.; Cotruvo, J.A., Jr. Iron-responsive riboswitches. Curr. Opin. Chem. Biol. 2022, 68, 102135. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.A.; Frederick, T.E.; Mallimadugula, U.L.; Singh, S.; Vithani, N.; Zimmerman, M.I.; Porter, J.R.; Moeder, K.E.; Amarasinghe, G.K.; Bowman, G.R. A cryptic pocket in Ebola VP35 allosterically controls RNA binding. Nat. Commun. 2022, 13, 2269. [Google Scholar] [CrossRef]
- Schroeder, G.M.; Cavender, C.E.; Blau, M.E.; Jenkins, J.L.; Mathews, D.H.; Wedekind, J.E. A small RNA that cooperatively senses two stacked metabolites in one pocket for gene control. Nat. Commun. 2022, 13, 199. [Google Scholar] [CrossRef]
- Petushkov, I.; Pupov, D.; Bass, I.; Kulbachinskiy, A. Mutations in the CRE pocket of bacterial RNA polymerase affect multiple steps of transcription. Nucleic Acids Res. 2015, 43, 5798–5809. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Y. Integrated modeling of protein and RNA. Brief. Bioinform. 2024, 25, bbae139. [Google Scholar] [CrossRef]
- Vicens, Q.; Mondragon, E.; Batey, R.T. Molecular sensing by the aptamer domain of the FMN riboswitch: A general model for ligand binding by conformational selection. Nucleic Acids Res. 2011, 39, 8586–8598. [Google Scholar] [CrossRef]
- Yang, R.; Liu, H.; Yang, L.; Zhou, T.; Li, X.; Zhao, Y. RPpocket: An RNA-Protein Intuitive Database with RNA Pocket Topology Resources. Int. J. Mol. Sci. 2022, 23, 6903. [Google Scholar] [CrossRef]
- Wang, H.W.; Wang, K.L.; Guan, Z.Y.; Jian, Y.R.; Jia, Y.; Kashanchi, F.; Zeng, C.; Zhao, Y.J. Computational study of non-catalytic T-loop pocket on CDK proteins for drug development. Chin. Phys. B 2017, 26, 128702. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, J.; Liu, H.; Xu, Y.; Jia, Y.; Zhao, Y. HKPocket: Human kinase pocket database for drug design. BMC Bioinform. 2019, 20, 617. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, W.M.; Calabrese, D.R.; Schneekloth, J.S., Jr. Evidence for ligandable sites in structured RNA throughout the Protein Data Bank. Bioorg. Med. Chem. 2019, 27, 2253–2260. [Google Scholar] [CrossRef]
- Pan, Z.; Zhou, S.; Liu, T.; Liu, C.; Zang, M.; Wang, Q. WVDL: Weighted Voting Deep Learning Model for Predicting RNA-Protein Binding Sites. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 3322–3328. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Time | Description | Data Availability | Reference |
|---|---|---|---|---|
| SMMRNA | 2014 | The database of small molecule modulators along with their target RNA and experimentally determined binding data. | N/A | [14] |
| NALDB | 2016 | The database provides detailed information about the experimental data of small molecules that were reported to target all types of nucleic acid structures. | https://www.iiti.ac.in/people/~amitk/bsbe/naldb/HOME.php (9 December 2024) | [15] |
| Inforna 2.0 | 2017 | A small molecule design platform for structured RNAs that integrates all known RNA motif–small molecule binding partners reported in the scientific literature. | N/A | [44] |
| NoncoRNA | 2020 | A manually curated database of ncRNAs and drug target associations designed to provide a potential resource of high-quality data for the exploration of drug sensitivity-related ncRNAs in a variety of human cancers. | http://www.ncdtcdb.cn:8080/NoncoRNA/ (9 December 2024) | [39] |
| RPocket | 2021 | An intuitive database of RNA pocket topology information with RNA–ligand data resources that provides geometrical size, centroid, shape, and secondary structure elements of RNA pockets. | http://zhaoserver.com.cn/RPocket/RPocket.html (9 December 2024) | [47] |
| R-BIND | 2022 | An updated database of bioactive RNA-targeting small molecules and associated RNA secondary structures. | https://rbind.chem.duke.edu (9 December 2024) | [45] |
| RNALigands | 2022 | A database of RNA secondary structure motifs and small molecular ligands. | https://github.com/SaisaiSun/RNALigands (9 December 2024) | [43] |
| HARIBOSS | 2022 | A database on the pocket structure and physicochemical properties of ligands and RNA. | http://hariboss.pasteur.cloud (9 December 2024) | [46] |
| R-SIM | 2023 | An experimentally validated database of RNA–small molecule interactions that provides comprehensive information on the sequence, structure, and classification of RNA, various physicochemical properties of small molecules, binding affinities, and the literature sources of the data. | https://web.iitm.ac.in/bioinfo2/R_SIM/index.html (9 December 2024) | [42] |
| ROBIN | 2023 | A library of nucleic acid binders was identified by small molecule microarray (SMM) screening that reported 2003 RNA–ligand small molecules, representing the largest fully publicly available experimentally derived library to date. | https://github.com/ky66/ROBIN (9 December 2024) | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, C.; Zeng, C.; Liu, H.; Wang, H.; Peng, Y.; Zhao, Y. Advances and Mechanisms of RNA–Ligand Interaction Predictions. Life 2025, 15, 104. https://doi.org/10.3390/life15010104
Zhuo C, Zeng C, Liu H, Wang H, Peng Y, Zhao Y. Advances and Mechanisms of RNA–Ligand Interaction Predictions. Life. 2025; 15(1):104. https://doi.org/10.3390/life15010104
Chicago/Turabian StyleZhuo, Chen, Chengwei Zeng, Haoquan Liu, Huiwen Wang, Yunhui Peng, and Yunjie Zhao. 2025. "Advances and Mechanisms of RNA–Ligand Interaction Predictions" Life 15, no. 1: 104. https://doi.org/10.3390/life15010104
APA StyleZhuo, C., Zeng, C., Liu, H., Wang, H., Peng, Y., & Zhao, Y. (2025). Advances and Mechanisms of RNA–Ligand Interaction Predictions. Life, 15(1), 104. https://doi.org/10.3390/life15010104