
Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer


 and
and
Abstract
1. Introduction
2. PTMs’ Role in Cancer Hallmarks and Enabling Characteristics
2.1. PTMs and Cancer Hallmarks
2.1.1. Sustaining Proliferative Signaling
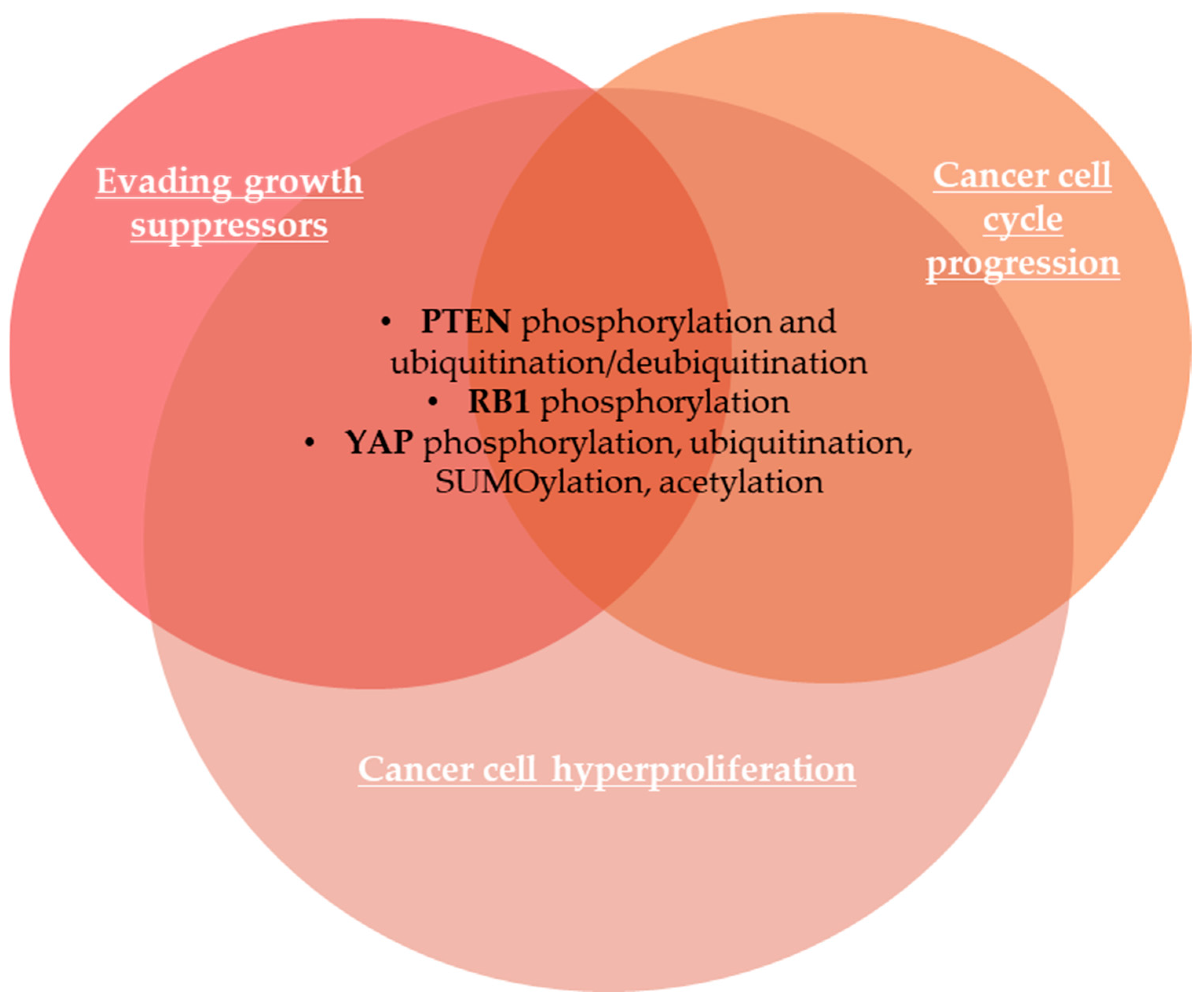
2.1.2. Evading Growth Suppressors
2.1.3. Avoiding Immune Destruction
2.1.4. Enabling Replicative Immortality
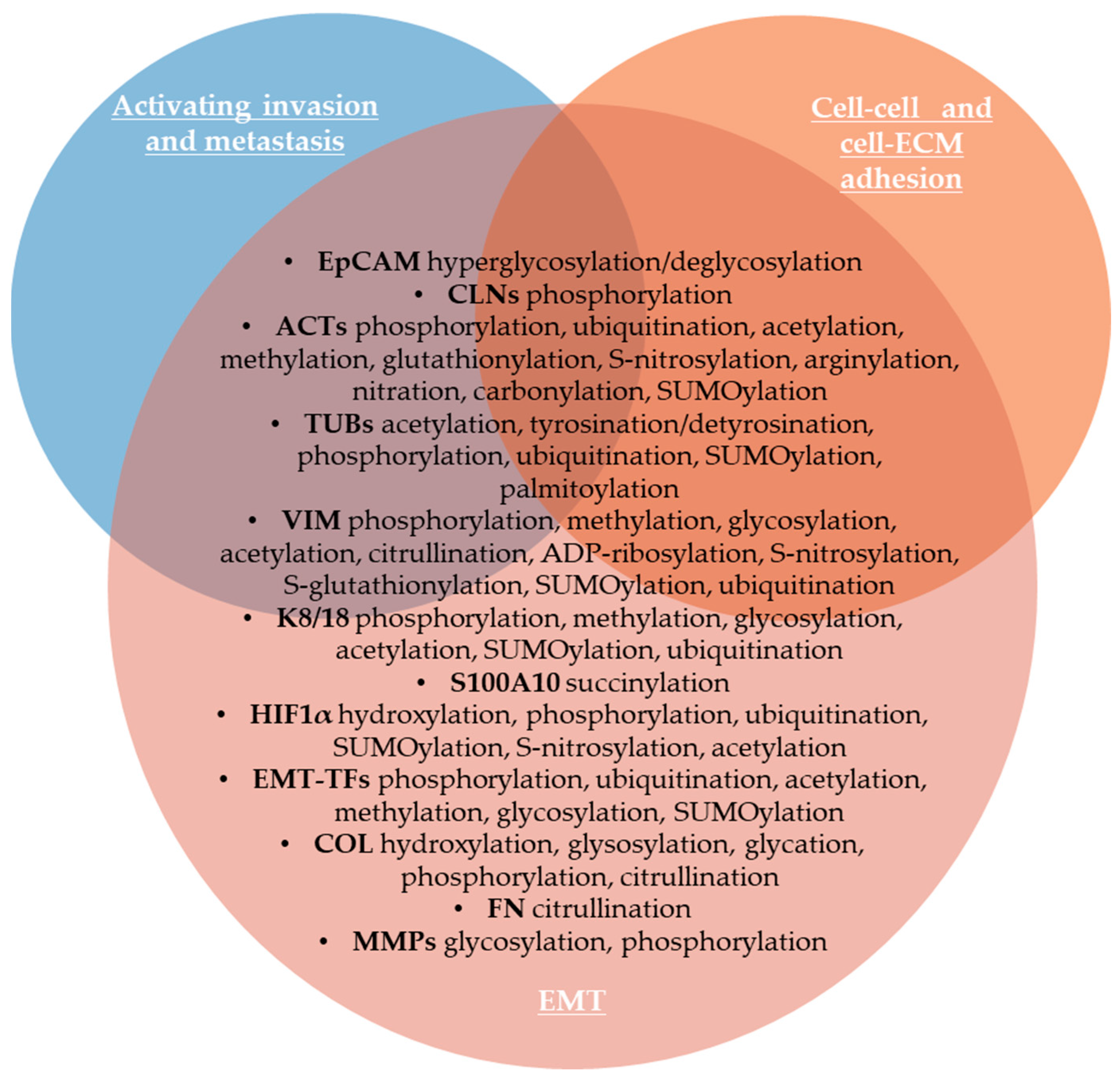
2.1.5. Activating Invasion and Metastasis
2.1.6. Inducing or Accessing Vasculature
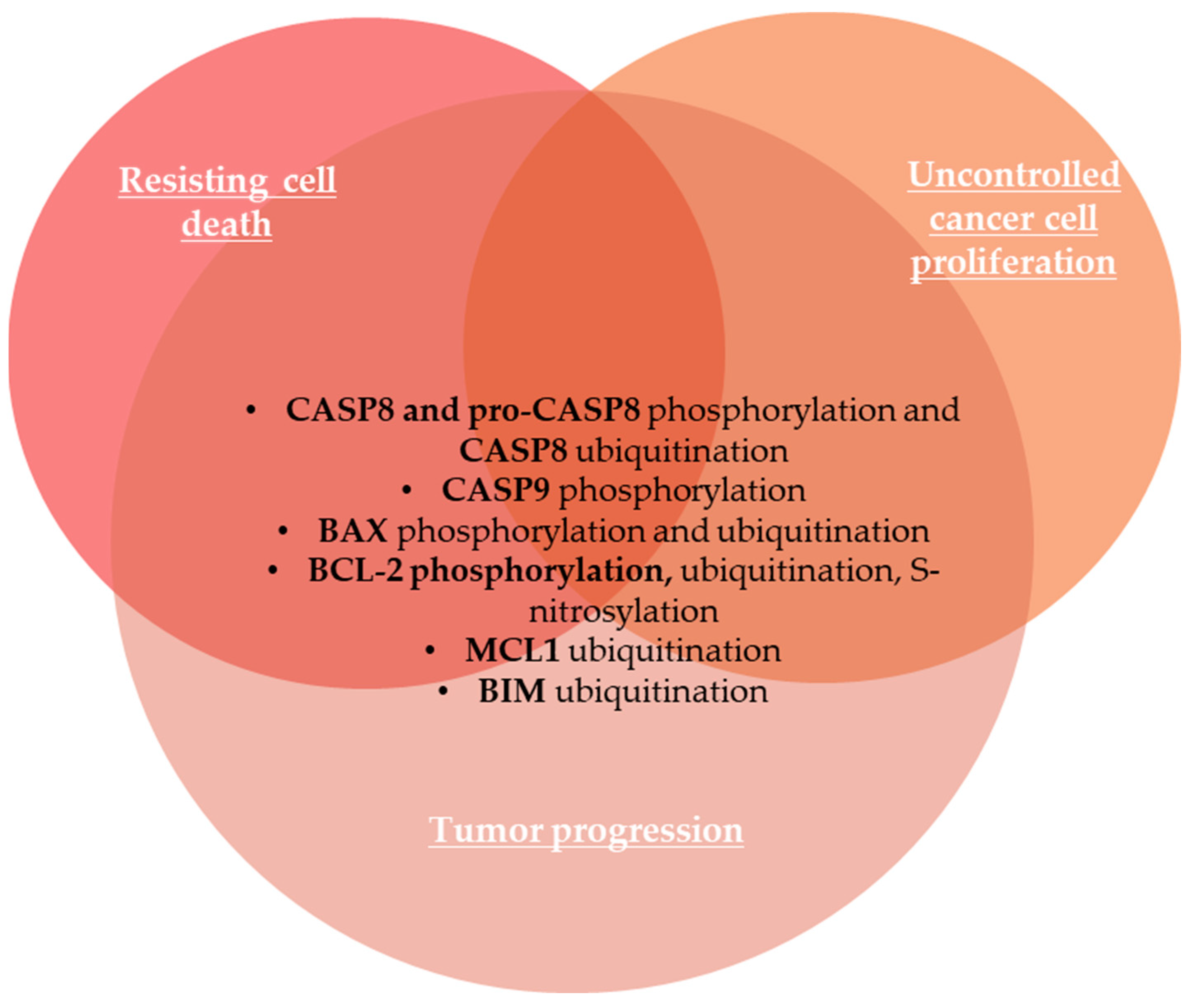
2.1.7. Resisting Cell Death
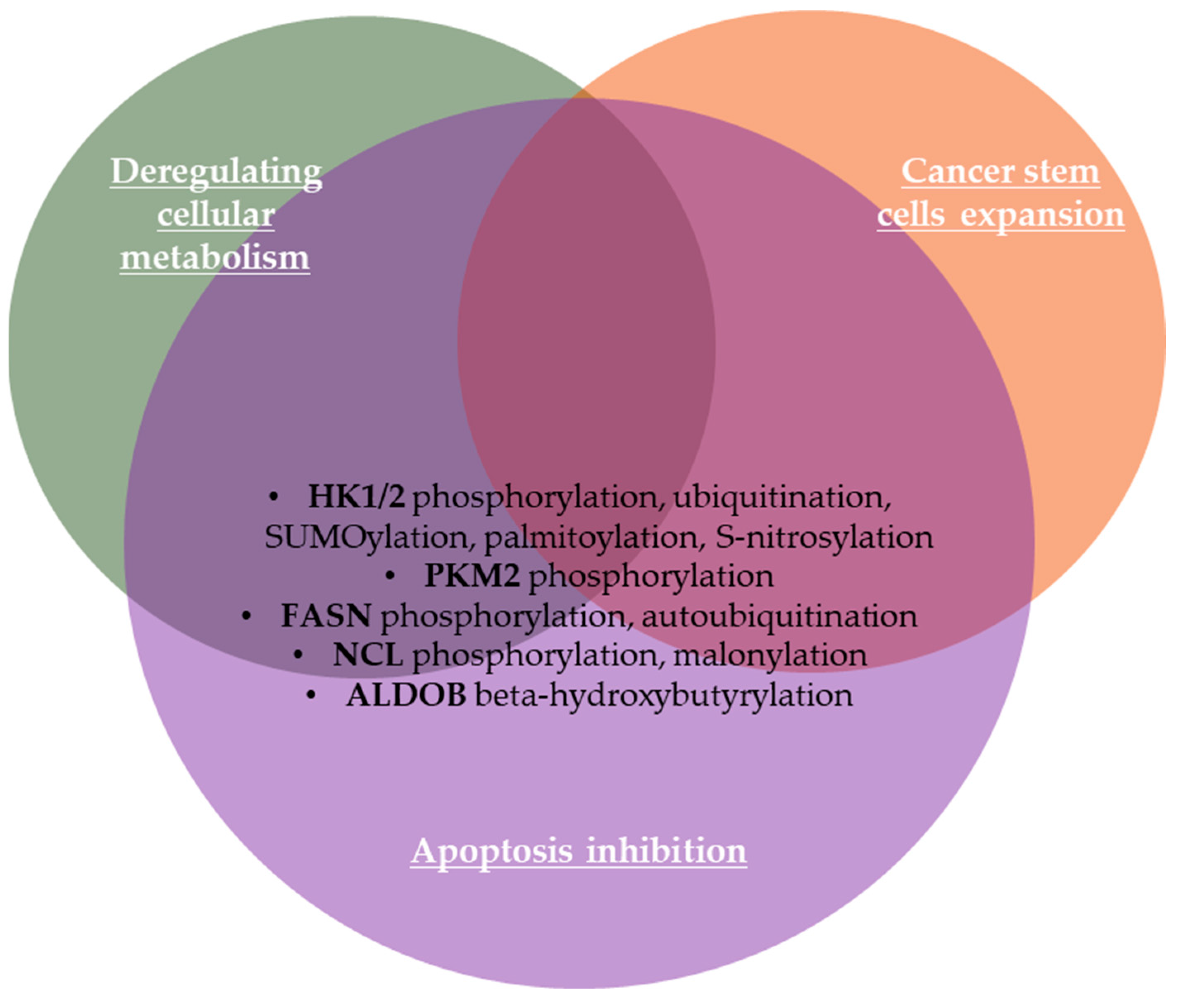
2.1.8. Deregulating Cellular Metabolism
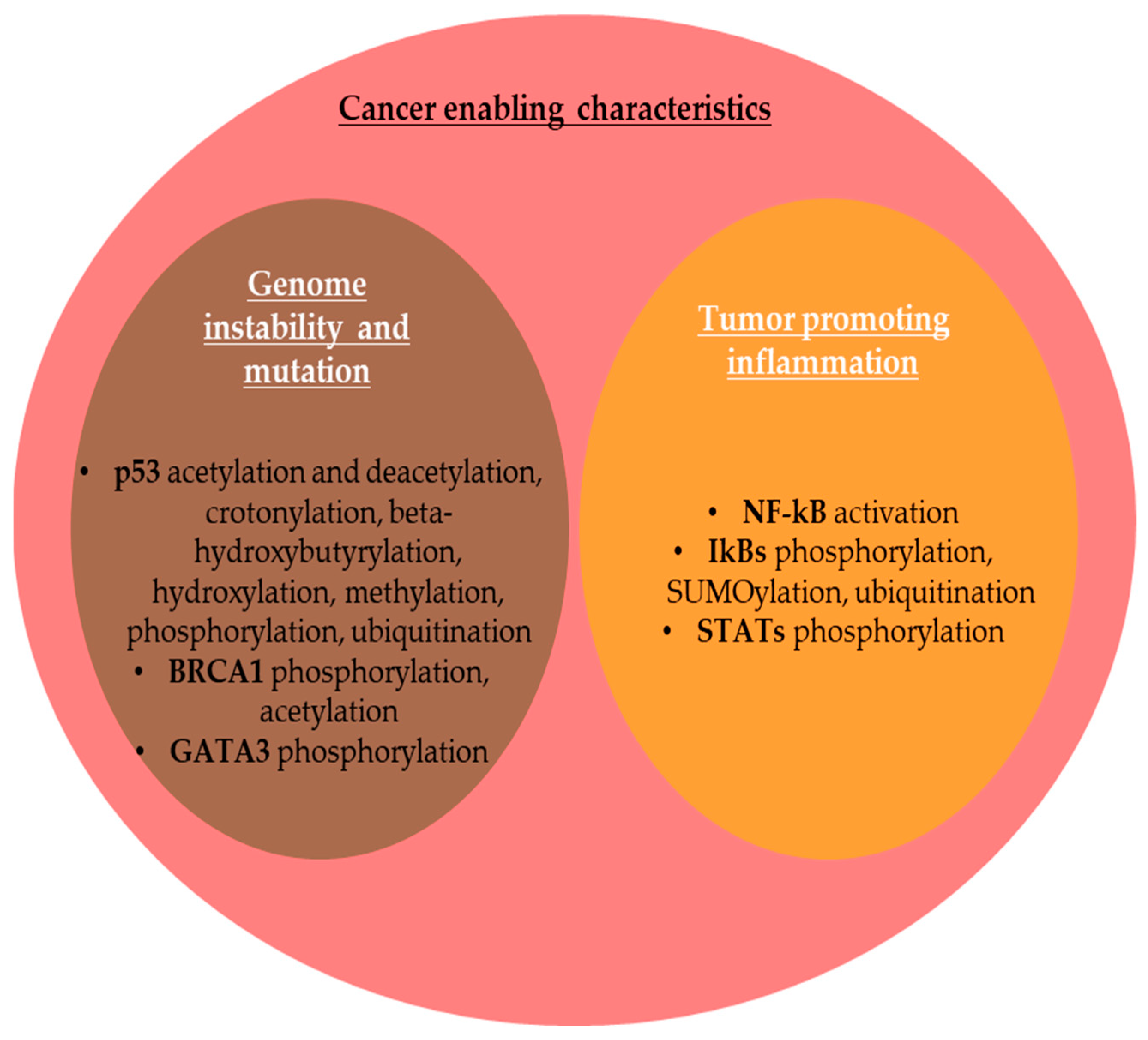
2.2. PTMs and Cancer Enabling Characteristics
2.2.1. Genome Instability and Mutation
2.2.2. Tumor-Promoting Inflammation
2.3. PTMs Involved in Additional Emerging Hallmarks and Enabling Characteristics of Cancer
2.3.1. Unlocking Phenotypic Plasticity
2.3.2. Nonmutational Epigenetic Reprogramming
2.3.3. Polymorphic Microbiomes
2.3.4. Senescent Cells
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aranda-Anzaldo, A.; Dent, M.A.R. Is cancer a disease set up by cellular stress responses? Cell Stress Chaperones 2021, 26, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Y.; Jian, Y.; Gu, L.; Zhang, D.; Zhou, H.; Wang, Y.; Xu, Z.-X. The regulations of telomerase reverse transcriptase (TERT) in cancer. Cell Death Dis. 2024, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.; Brooks, J.; Batis, N.; Khan, N.; El-Asrag, M.; Nankivell, P.; Mehanna, H.; Taylor, G. Feasibility of mass cytometry proteomic characterisation of circulating tumour cells in head and neck squamous cell carcinoma for deep phenotyping. Br. J. Cancer 2023, 129, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Rein, T. Post-translational modifications and stress adaptation: The paradigm of FKBP51. Biochem. Soc. Trans. 2020, 48, 441–449. [Google Scholar] [CrossRef]
- Suskiewicz, M.J. The logic of protein post-translational modifications (PTMs): Chemistry, mechanisms and evolution of protein regulation through covalent attachments. BioEssays 2024, 46, 2300178. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Prem Kumar, A.; Seok Ahn, K.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2017, 9, 11414–11426. [Google Scholar] [CrossRef]
- Brentville, V.A.; Metheringham, R.L.; Gunn, B.; Symonds, P.; Daniels, I.; Gijon, M.; Cook, K.; Xue, W.; Durrant, L.G. Citrullinated Vimentin Presented on MHC-II in Tumor Cells Is a Target for CD4+ T-Cell–Mediated Antitumor Immunity. Cancer Res. 2016, 76, 548–560. [Google Scholar] [CrossRef]
- Dutta, H.; Jain, N. Post-translational modifications and their implications in cancer. Front. Oncol. 2023, 13, 1240115. [Google Scholar] [CrossRef]
- Ko, P.J.; Dixon, S.J. Protein palmitoylation and cancer. EMBO Rep. 2018, 19, e46666. [Google Scholar] [CrossRef]
- Madzharova, E.; Kastl, P.; Sabino, F.; auf dem Keller, U. Post-Translational Modification-Dependent Activity of Matrix Metalloproteinases. Int. J. Mol. Sci. 2019, 20, 3077. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Ahmed, R.; Ahmed, H.; Chatterjee, B.P. Phosphorylated Proteins from Serum: A Promising Potential Diagnostic Biomarker of Cancer. Int. J. Mol. Sci. 2022, 23, 12359. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Shen, D.; Ye, J.; Chen, J.; Li, J. PhosCancer: A comprehensive database for investigating protein phosphorylation in human cancer. iScience 2024, 27, 111060. [Google Scholar] [CrossRef] [PubMed]
- Landré, V.; Revi, B.; Mir, M.G.; Verma, C.; Hupp, T.R.; Gilbert, N.; Ball, K.L. Regulation of transcriptional activators by DNA-binding domain ubiquitination. Cell Death Differ. 2017, 24, 903–916. [Google Scholar] [CrossRef]
- Tang, X.; Sui, X.; Weng, L.; Liu, Y. SNAIL1: Linking Tumor Metastasis to Immune Evasion. Front. Immunol. 2021, 12, 724200. [Google Scholar] [CrossRef]
- Rosonina, E.; Akhter, A.; Dou, Y.; Babu, J.; Sri Theivakadadcham, V.S. Regulation of transcription factors by sumoylation. Transcription 2017, 8, 220–231. [Google Scholar] [CrossRef]
- Yang, J.; Song, C.; Zhan, X. The role of protein acetylation in carcinogenesis and targeted drug discovery. Front. Endocrinol. 2022, 13, 972312. [Google Scholar] [CrossRef]
- López-Bañuelos, L.; Vega, L. Inhibition of acetylation, is it enough to fight cancer? Crit. Rev. Oncol./Hematol. 2022, 176, 103752. [Google Scholar] [CrossRef]
- Shi, H.; Cui, W.; Qin, Y.; Chen, L.; Yu, T.; Lv, J. A glimpse into novel acylations and their emerging role in regulating cancer metastasis. Cell. Mol. Life Sci. 2024, 81, 76. [Google Scholar] [CrossRef]
- Sun, L.; Meng, H.; Liu, T.; Zhao, Q.; Xia, M.; Zhao, Z.; Qian, Y.; Cui, H.; Zhong, X.; Chai, K.; et al. Nucleolin malonylation as a nuclear-cytosol signal exchange mechanism to drive cell proliferation in Hepatocarcinoma by enhancing AKT translation. J. Biol. Chem. 2024, 300, 107785. [Google Scholar] [CrossRef]
- Yang, P.; Qin, Y.; Zeng, L.; He, Y.; Xie, Y.; Cheng, X.; Huang, W.; Cao, L. Crotonylation and disease: Current progress and future perspectives. Biomed. Pharmacother. 2023, 165, 115108. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Han, D. A review of the mechanism of succinylation in cancer. Medicine 2022, 101, e31493. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Huang, X.; Gou, S.; Zhang, S.; Gou, Y.; Zhang, Q.; Chen, H.; Sun, L.; Chen, M.; Liu, D.; et al. Ketogenic diet reshapes cancer metabolism through lysine β-hydroxybutyrylation. Nat. Metab. 2024, 6, 1505–1528. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, D.; Weng, Y.; Delaney, K.; Tang, Z.; Yan, C.; Qi, S.; Peng, C.; Cole, P.A.; Roeder, R.G.; et al. The regulatory enzymes and protein substrates for the lysine β-hydroxybutyrylation pathway. Sci. Adv. 2021, 7, eabe2771. [Google Scholar] [CrossRef]
- Shang, S.; Liu, J.; Hua, F. Protein acylation: Mechanisms, biological functions and therapeutic targets. Signal Transduct. Target. Ther. 2022, 7, 396. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2016, 1864, 1372–1401. [Google Scholar] [CrossRef]
- Liang, D.; Gao, Q.; Meng, Z.; Li, W.; Song, J.; Xue, K. Glycosylation in breast cancer progression and mammary development: Molecular connections and malignant transformations. Life Sci. 2023, 326, 121781. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, Q.; Li, Z.; Zhao, Y.; Sun, Y. Protein neddylation and its role in health and diseases. Signal Transduct. Target. Ther. 2024, 9, 85. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Katayama, H.; Kobayashi, M.; Irajizad, E.; Sevillano, A.M.; Patel, N.; Mao, X.; Rusling, L.; Vykoukal, J.; Cai, Y.; Hsiao, F.; et al. Protein citrullination as a source of cancer neoantigens. J. Immunother. Cancer 2021, 9, e002549. [Google Scholar] [CrossRef]
- Singh, V.; Ram, M.; Kumar, R.; Prasad, R.; Roy, B.K.; Singh, K.K. Phosphorylation: Implications in Cancer. Protein J. 2017, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Wang, Y.; Yang, M.; Hong, F.; Yang, S. Protein Phosphorylation in Cancer: Role of Nitric Oxide Signaling Pathway. Biomolecules 2021, 11, 1009. [Google Scholar] [CrossRef] [PubMed]
- Coopman, P. Protein Phosphorylation in Cancer: Unraveling the Signaling Pathways. Biomolecules 2022, 12, 1036. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ren, T.; Zhang, Y.; Nan, N. Methylation multiplicity and its clinical values in cancer. Expert Rev. Mol. Med. 2021, 23, e2. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Li, K.; Li, H.; Zhu, Y.; Zhai, Y.; Lu, B.; Fan, Y.; Liu, Z.; Chen, X.; et al. Ubiquitination and deubiquitination in cancer: From mechanisms to novel therapeutic approaches. Mol. Cancer 2024, 23, 148. [Google Scholar] [CrossRef]
- Dagar, G.; Kumar, R.; Yadav, K.K.; Singh, M.; Pandita, T.K. Ubiquitination and deubiquitination: Implications on cancer therapy. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2023, 1866, 194979. [Google Scholar] [CrossRef]
- Sampson, C.; Wang, Q.; Otkur, W.; Zhao, H.; Lu, Y.; Liu, X.; Piao, H.-l. The roles of E3 ubiquitin ligases in cancer progression and targeted therapy. Clin. Transl. Med. 2023, 13, e1204. [Google Scholar] [CrossRef]
- Gu, Y.; Fang, Y.; Wu, X.; Xu, T.; Hu, T.; Xu, Y.; Ma, P.; Wang, Q.; Shu, Y. The emerging roles of SUMOylation in the tumor microenvironment and therapeutic implications. Exp. Hematol. Oncol. 2023, 12, 58. [Google Scholar] [CrossRef]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Liu, D.; Che, X.; Wu, G. Deciphering the role of neddylation in tumor microenvironment modulation: Common outcome of multiple signaling pathways. Biomark. Res. 2024, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Best, S.; Danilov, A.V. Neddylation and anti-tumor immunity. Oncotarget 2021, 12, 2227–2230. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Yu, C.; Deng, X.; Zhang, Y.; Zhang, X.; Duan, F. O-GlcNAcylation and Its Role in Cancer-Associated Inflammation. Front. Immunol. 2022, 13, 861559. [Google Scholar] [CrossRef]
- Cao, D.; Sun, W.; Li, X.; Jian, L.; Zhou, X.; Bode, A.M.; Luo, X. The role of novel protein acylations in cancer. Eur. J. Pharmacol. 2024, 979, 176841. [Google Scholar] [CrossRef]
- Jeong, K.; Choi, J.; Choi, A.; Shim, J.; Kim, Y.; Oh, C.; Youn, H.-D.; Cho, E.-J. Determination of HIF-1α degradation pathways via modulation of the propionyl mark. BMB Rep. 2023, 56, 252–257. [Google Scholar] [CrossRef]
- Yang, S.; Fan, X.; Yu, W. Regulatory Mechanism of Protein Crotonylation and Its Relationship with Cancer. Cells 2024, 13, 1812. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Liang, Z.; Meng, Z.; Zhang, X.; Ma, G.; Chen, Y.; Zhang, M.; Su, Y.; Li, Z.; et al. Modification of lysine-260 2-hydroxyisobutyrylation destabilizes ALDH1A1 expression to regulate bladder cancer progression. iScience 2023, 26, 108142. [Google Scholar] [CrossRef]
- Hong, G.; Su, X.; Xu, K.; Liu, B.; Wang, G.; Li, J.; Wang, R.; Zhu, M.; Li, G. Salt stress downregulates 2-hydroxybutyrylation in Arabidopsis siliques. J. Proteom. 2022, 250, 104383. [Google Scholar] [CrossRef]
- Zheng, H.; Mei, H.; Li, X.; Li, D.; Liu, W. Proteome-Wide Analysis of Lysine 2-Hydroxyisobutyrylation in Aspergillus fumigatus. Curr. Microbiol. 2024, 81, 74. [Google Scholar] [CrossRef]
- Li, H.; Sun, L.; Gao, P.; Hu, H. Lactylation in cancer: Current understanding and challenges. Cancer Cell 2024, 42, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Zhang, J.; Lu, J.; Zhang, G.; Hua, M.; Guo, W.; Yang, J.; Fan, G. A review of lactate-lactylation in malignancy: Its potential in immunotherapy. Front. Immunol. 2024, 15, 1384948. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhou, Y.; Xue, Z.; Hao, N.; Li, Y.; Guo, X.; Wang, D.; Shi, X.; Li, H. Histone benzoylation serves as an epigenetic mark for DPF and YEATS family proteins. Nucleic Acids Res. 2020, 49, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Greaves, J.; Chamberlain, L.H. New links between S-acylation and cancer. J. Pathol. 2014, 233, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E. Citrullination in Cancer. Cancer Res. 2019, 79, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhang, Y.; Wang, S. Histone citrullination: A new target for tumors. Mol. Cancer 2021, 20, 90. [Google Scholar] [CrossRef]
- Rappu, P.; Suwal, U.; Siljamäki, E.; Heino, J. Inflammation-related citrullination of matrisome proteins in human cancer. Front. Oncol. 2022, 12, 1035188. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Filippopoulou, C.; Simos, G.; Chachami, G. The Role of Sumoylation in the Response to Hypoxia: An Overview. Cells 2020, 9, 2359. [Google Scholar] [CrossRef]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Cell Physiol. 2015, 309, C775–C782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, M. Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers. Int. J. Mol. Sci. 2023, 24, 9173. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, J.; Russell, J.D.; Yu, P.; Westphall, M.S.; Coon, J.J.; Thomson, J.A. NANOG Is Multiply Phosphorylated and Directly Modified by ERK2 and CDK1 In Vitro. Stem Cell Rep. 2014, 2, 18–25. [Google Scholar] [CrossRef]
- Ooki, A.; VandenBussche, C.J.; Kates, M.; Hahn, N.M.; Matoso, A.; McConkey, D.J.; Bivalacqua, T.J.; Hoque, M.O. CD24 regulates cancer stem cell (CSC)-like traits and a panel of CSC-related molecules serves as a non-invasive urinary biomarker for the detection of bladder cancer. Br. J. Cancer 2018, 119, 961–970. [Google Scholar] [CrossRef]
- Pallegar, N.K.; Ayre, D.C.; Christian, S.L. Repression of CD24 surface protein expression by oncogenic Ras is relieved by inhibition of Raf but not MEK or PI3K. Front. Cell Dev. Biol. 2015, 3, 47. [Google Scholar] [CrossRef]
- Skowron, M.A.; Becker, T.K.; Kurz, L.; Jostes, S.; Bremmer, F.; Fronhoffs, F.; Funke, K.; Wakileh, G.A.; Müller, M.R.; Burmeister, A.; et al. The signal transducer CD24 suppresses the germ cell program and promotes an ectodermal rather than mesodermal cell fate in embryonal carcinomas. Mol. Oncol. 2022, 16, 982–1008. [Google Scholar] [CrossRef]
- Shen, Y.-A.; Wang, C.-Y.; Chuang, H.-Y.; Hwang, J.J.-J.; Chi, W.-H.; Shu, C.-H.; Ho, C.-Y.; Li, W.-Y.; Chen, Y.-J. CD44 and CD24 coordinate the reprogramming of nasopharyngeal carcinoma cells towards a cancer stem cell phenotype through STAT3 activation. Oncotarget 2016, 7, 58351–58366. [Google Scholar] [CrossRef]
- Liao, C.; Wang, Q.; An, J.; Chen, J.; Li, X.; Long, Q.; Xiao, L.; Guan, X.; Liu, J. CD44 Glycosylation as a Therapeutic Target in Oncology. Front. Oncol. 2022, 12, 883831. [Google Scholar] [CrossRef]
- Deng, L.; Zhang, X.; Xiang, X.; Xiong, R.; Xiao, D.; Chen, Z.; Liu, K.; Feng, G. NANOG Promotes Cell Proliferation, Invasion, and Stemness via IL-6/STAT3 Signaling in Esophageal Squamous Carcinoma. Technol. Cancer Res. Treat. 2021, 20, 153303382110384. [Google Scholar] [CrossRef]
- Liu, X.; Yao, Y.; Ding, H.; Han, C.; Chen, Y.; Zhang, Y.; Wang, C.; Zhang, X.; Zhang, Y.; Zhai, Y.; et al. USP21 deubiquitylates Nanog to regulate protein stability and stem cell pluripotency. Signal Transduct. Target. Ther. 2016, 1, 16024. [Google Scholar] [CrossRef] [PubMed]
- Valverde, J.M.; Dubra, G.; Phillips, M.; Haider, A.; Elena-Real, C.; Fournet, A.; Alghoul, E.; Chahar, D.; Andrés-Sanchez, N.; Paloni, M.; et al. A cyclin-dependent kinase-mediated phosphorylation switch of disordered protein condensation. Nat. Commun. 2023, 14, 6316. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Khoshbakht, T.; Hussen, B.M.; Dong, P.; Gassler, N.; Taheri, M.; Baniahmad, A.; Dilmaghani, N.A. A review on the role of cyclin dependent kinases in cancers. Cancer Cell Int. 2022, 22, 325. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Pei, C.; Ouyang, M.; Chen, Y.; Yu, L.; Huang, X.; Tan, Y. The antitumor peptide M1-20 induced the degradation of CDK1 through CUL4-DDB1-DCAF1-involved ubiquitination. Cancer Gene Ther. 2024. [Google Scholar] [CrossRef]
- Remnant, L.; Kochanova, N.Y.; Reid, C.; Cisneros-Soberanis, F.; Earnshaw, W.C. The intrinsically disorderly story of Ki-67. Open Biol. 2021, 11, 210120. [Google Scholar] [CrossRef]
- Massacci, G.; Perfetto, L.; Sacco, F. The Cyclin-dependent kinase 1: More than a cell cycle regulator. Br. J. Cancer 2023, 129, 1707–1716. [Google Scholar] [CrossRef]
- Myatt, S.S.; Kongsema, M.; Man, C.W.Y.; Kelly, D.J.; Gomes, A.R.; Khongkow, P.; Karunarathna, U.; Zona, S.; Langer, J.K.; Dunsby, C.W.; et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene 2014, 33, 4316–4329. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Guillen, V.S.; Katzenellenbogen, J.A. Targeting the oncogenic transcription factor FOXM1 to improve outcomes in all subtypes of breast cancer. Breast Cancer Res. 2023, 25, 76. [Google Scholar] [CrossRef]
- Lin, X.; He, Y.; Liu, Y.; Zhou, H.; Xu, X.; Xu, J.; Zhou, K. CDK1 promotes the phosphorylation of KIFC1 to regulate the tumorgenicity of endometrial carcinoma. J. Gynecol. Oncol. 2024, 35, e68. [Google Scholar] [CrossRef]
- Davey, M.G.; Hynes, S.O.; Kerin, M.J.; Miller, N.; Lowery, A.J. Ki-67 as a Prognostic Biomarker in Invasive Breast Cancer. Cancers 2021, 13, 4455. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, J.; Zhang, Q.-W.; Zhao, W.; Guo, J.-H.; Liu, S.-L.; Wu, Y.-L.; Jiang, B.; Gao, F.-H. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int. J. Biochem. Cell Biol. 2016, 79, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Endl, E.; Gerdes, J. Posttranslational modifications of the KI-67 protein coincide with two major checkpoints during mitosis. J. Cell. Physiol. 2000, 182, 371–380. [Google Scholar] [CrossRef]
- Tan, N.Y.; Khachigian, L.M. Sp1 Phosphorylation and Its Regulation of Gene Transcription. Mol. Cell. Biol. 2009, 29, 2483–2488. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Wang, S.A.; Yang, W.B.; Yang, H.C.; Hung, C.Y.; Su, T.P.; Chang, W.C.; Hung, J.J. Sp1 phosphorylation by cyclin-dependent kinase 1/cyclin B1 represses its DNA-binding activity during mitosis in cancer cells. Oncogene 2012, 31, 4946–4959. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.M.R.; Karpowicz, P.A.; Carey, T.E.; Arbiser, J.; Nahta, R.; Chen, Z.G.; Dong, J.-T.; Kucuk, O.; Khan, G.N.; Huang, G.S.; et al. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin. Cancer Biol. 2015, 35, S55–S77. [Google Scholar] [CrossRef]
- Dempsey, D.R.; Viennet, T.; Iwase, R.; Park, E.; Henriquez, S.; Chen, Z.; Jeliazkov, J.R.; Palanski, B.A.; Phan, K.L.; Coote, P.; et al. The structural basis of PTEN regulation by multi-site phosphorylation. Nat. Struct. Mol. Biol. 2021, 28, 858–868. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Xia, Q.; Ali, S.; Liu, L.; Li, Y.; Liu, X.; Zhang, L.; Dong, L. Role of Ubiquitination in PTEN Cellular Homeostasis and Its Implications in GB Drug Resistance. Front. Oncol. 2020, 10, 1569. [Google Scholar] [CrossRef]
- Wang, K.; Liu, J.; Li, Y.-L.; Li, J.-P.; Zhang, R. Ubiquitination/de-ubiquitination: A promising therapeutic target for PTEN reactivation in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188723. [Google Scholar] [CrossRef]
- Vélez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776. [Google Scholar] [CrossRef]
- Matsui, T.; Nieto-Estévez, V.; Kyrychenko, S.; Schneider, J.W.; Hsieh, J. Retinoblastoma protein controls growth, survival and neuronal migration in human cerebral organoids. Development 2017, 144, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.; Mason-Richie, N.A.; Gettler, C.A.; Wikenheiser-Brokamp, K.A. Retinoblastoma Family Proteins Have Distinct Functions in Pulmonary Epithelial Cells In vivo Critical for Suppressing Cell Growth and Tumorigenesis. Cancer Res. 2009, 69, 8733–8741. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, W.; Li, Z.; Lu, J.; Wang, X. The interplay between p16 serine phosphorylation and arginine methylation determines its function in modulating cellular apoptosis and senescence. Sci. Rep. 2017, 7, 41390. [Google Scholar] [CrossRef] [PubMed]
- Nita, A.; Moroishi, T. Hippo pathway in cell–cell communication: Emerging roles in development and regeneration. Inflamm. Regen. 2024, 44, 18. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. [Google Scholar] [CrossRef]
- Yan, F.; Qian, M.; He, Q.; Zhu, H.; Yang, B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2020, 1864, 129397. [Google Scholar] [CrossRef]
- Luo, J.; Zou, H.; Guo, Y.; Tong, T.; Chen, Y.; Xiao, Y.; Pan, Y.; Li, P. The oncogenic roles and clinical implications of YAP/TAZ in breast cancer. Br. J. Cancer 2023, 128, 1611–1624. [Google Scholar] [CrossRef]
- Lin, X.; Kang, K.; Chen, P.; Zeng, Z.; Li, G.; Xiong, W.; Yi, M.; Xiang, B. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 2024, 23, 108. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Duan, Z.; Shi, R.; Gao, B.; Cai, J. N-linked glycosylation of PD-L1/PD-1: An emerging target for cancer diagnosis and treatment. J. Transl. Med. 2024, 22, 705. [Google Scholar] [CrossRef]
- Wang, R.; He, S.; Long, J.; Wang, Y.; Jiang, X.; Chen, M.; Wang, J. Emerging therapeutic frontiers in cancer: Insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp. Hematol. Oncol. 2024, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, J.; Chu, M.; Liu, Y.; Wang, Z.-W.; Zhu, X. Emerging Role of Ubiquitination in the Regulation of PD-1/PD-L1 in Cancer Immunotherapy. Mol. Ther. 2021, 29, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Lee, H.-H.; Hsu, J.L.; Yu, D.; Hung, M.-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Shu, Z.; Dwivedi, B.; Switchenko, J.M.; Yu, D.S.; Deng, X. PD-L1 deglycosylation promotes its nuclear translocation and accelerates DNA double-strand-break repair in cancer. Nat. Commun. 2024, 15, 6830. [Google Scholar] [CrossRef]
- Zhou, S.; Zhao, X.; Yang, Z.; Yang, R.; Chen, C.; Zhao, K.; Wang, W.; Ma, Y.; Zhang, Q.; Wang, X. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int. J. Cancer 2019, 145, 763–774. [Google Scholar] [CrossRef]
- Bailey, S.M. Editorial: Hallmark of cancer: Replicative immortality. Front. Oncol. 2023, 13, 1204094. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Razgonova, M.P.; Zakharenko, A.M.; Golokhvast, K.S.; Thanasoula, M.; Sarandi, E.; Nikolouzakis, K.; Fragkiadaki, P.; Tsoukalas, D.; Spandidos, D.A.; Tsatsakis, A. Telomerase and telomeres in aging theory and chronographic aging theory (Review). Mol. Med. Rep. 2020, 22, 1679–1694. [Google Scholar] [CrossRef]
- Shepelev, N.; Dontsova, O.; Rubtsova, M. Post-Transcriptional and Post-Translational Modifications in Telomerase Biogenesis and Recruitment to Telomeres. Int. J. Mol. Sci. 2023, 24, 5027. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Schiemann, W.P. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers 2022, 14, 808. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Mondal, P.; Sen, S.; Sengupta, S.; Das, C. SUMO E3 ligase CBX4 regulates hTERT-mediated transcription of CDH1 and promotes breast cancer cell migration and invasion. Biochem. J. 2020, 477, 3803–3818. [Google Scholar] [CrossRef]
- Xiao, D.; Xiong, M.; Wang, X.; Lyu, M.; Sun, H.; Cui, Y.; Chen, C.; Jiang, Z.; Sun, F. Regulation of the Function and Expression of EpCAM. Biomedicines 2024, 12, 1129. [Google Scholar] [CrossRef]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Zhan, Q.; Liu, B.; Situ, X.; Luo, Y.; Fu, T.; Wang, Y.; Xie, Z.; Ren, L.; Zhu, Y.; He, W.; et al. New insights into the correlations between circulating tumor cells and target organ metastasis. Signal Transduct. Target. Ther. 2023, 8, 465. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Sankpal, N.V.; Mayfield, J.D.; Willman, M.W.; Fleming, T.P.; Gillanders, W.E. Activator protein 1 (AP-1) contributes to EpCAM-dependent breast cancer invasion. Breast Cancer Res. 2011, 13, R124. [Google Scholar] [CrossRef]
- Gu, X.; Wei, S.; Lv, X. Circulating tumor cells: From new biological insights to clinical practice. Signal Transduct. Target. Ther. 2024, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Q.; Zhao, Q.; Yang, F.; Liu, T.; Huang, X.; Yan, Q.; Yang, X. Deglycosylated EpCAM regulates proliferation by enhancing autophagy of breast cancer cells via PI3K/Akt/mTOR pathway. Aging 2022, 14, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Sun, S.; Chen, Z.; Xiang, S.; Ding, Z.; Huang, Z.; Zhang, B. Understanding the versatile roles and applications of EpCAM in cancers: From bench to bedside. Exp. Hematol. Oncol. 2022, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, L.; Zhang, D.; Liu, T.; Yan, Q.; Yang, X. Deglycosylation of epithelial cell adhesion molecule affects epithelial to mesenchymal transition in breast cancer cells. J. Cell. Physiol. 2019, 234, 4504–4514. [Google Scholar] [CrossRef]
- Fagotto, F.; Aslemarz, A. EpCAM cellular functions in adhesion and migration, and potential impact on invasion: A critical review. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188436. [Google Scholar] [CrossRef]
- French, A.D.; Fiori, J.L.; Camilli, T.C.; Leotlela, P.D.; O’Connell, M.P.; Frank, B.P.; Subaran, S.; Indig, F.E.; Taub, D.D.; Weeraratna, A.T. PKC and PKA Phosphorylation Affect the Subcellular Localization of Claudin-1 in Melanoma Cells. Int. J. Med. Sci. 2009, 6, 93–101. [Google Scholar] [CrossRef]
- MacTaggart, B.; Kashina, A. Posttranslational modifications of the cytoskeleton. Cytoskeleton 2021, 78, 142–173. [Google Scholar] [CrossRef]
- Llorente-González, C.; González-Rodríguez, M.; Vicente-Manzanares, M. Targeting cytoskeletal phosphorylation in cancer. Explor. Target. Anti-Tumor Ther. 2021, 2, 292–308. [Google Scholar] [CrossRef]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Invadopodia: Clearing the way for cancer cell invasion. Ann. Transl. Med. 2020, 8, 902. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielińska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. BioMed Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef]
- Kuburich, N.A.; den Hollander, P.; Castaneda, M.; Pietilä, M.; Tang, X.; Batra, H.; Martínez-Peña, F.; Visal, T.H.; Zhou, T.; Demestichas, B.R.; et al. Stabilizing vimentin phosphorylation inhibits stem-like cell properties and metastasis of hybrid epithelial/mesenchymal carcinomas. Cell Rep. 2023, 42, 113470. [Google Scholar] [CrossRef] [PubMed]
- Berr, A.L.; Wiese, K.; dos Santos, G.; Koch, C.M.; Anekalla, K.R.; Kidd, M.; Davis, J.M.; Cheng, Y.; Hu, Y.-S.; Ridge, K.M. Vimentin is required for tumor progression and metastasis in a mouse model of non–small cell lung cancer. Oncogene 2023, 42, 2074–2087. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M.; McCulloch, C.A. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, Y.; Sui, Z.; Zhang, Y.; Liu, M.; Tang, H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 2016, 8, 48725–48736. [Google Scholar] [CrossRef]
- Fortier, A.-M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 Loss in Epithelial Cancer Cells Increases Collective Cell Migration and Cisplatin Sensitivity through Claudin1 Up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Neagu, A.-N.; Josan, C.-L.; Jayaweera, T.M.; Morrissiey, H.; Johnson, K.R.; Darie, C.C. Bio-Pathological Functions of Posttranslational Modifications of Histological Biomarkers in Breast Cancer. Molecules 2024, 29, 4156. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Li, X.; Shen, J.; Xu, Y.; Shi, H.; Mu, X.; Pan, J.; Zhao, T.; Li, M.; et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J. Cell. Mol. Med. 2019, 23, 293–305. [Google Scholar] [CrossRef]
- Shen, Z.; Yu, N.; Zhang, Y.; Jia, M.; Sun, Y.; Li, Y.; Zhao, L. The potential roles of HIF-1α in epithelial-mesenchymal transition and ferroptosis in tumor cells. Cell Signal. 2024, 122, 111345. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef]
- Fujisaki, H.; Futaki, S. Epithelial–Mesenchymal Transition Induced in Cancer Cells by Adhesion to Type I Collagen. Int. J. Mol. Sci. 2023, 24, 198. [Google Scholar] [CrossRef]
- Borst, R.; Meyaard, L.; Pascoal Ramos, M.I. Understanding the matrix: Collagen modifications in tumors and their implications for immunotherapy. J. Transl. Med. 2024, 22, 382. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Kerr, D.J.; Pezzella, F.; Ribatti, D. Accessing the vasculature in cancer: Revising an old hallmark. Trends Cancer 2024, 10, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Renault, M.-A.; Roncalli, J.; Tongers, J.; Thorne, T.; Klyachko, E.; Misener, S.; Volpert, O.V.; Mehta, S.; Burg, A.; Luedemann, C.; et al. Sonic hedgehog induces angiogenesis via Rho kinase-dependent signaling in endothelial cells. J. Mol. Cell. Cardiol. 2010, 49, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, K.; Shimo, T.; Murakami, J.; Anqi, C.; Kawai, H.; Yoshida, S.; Wathone Oo, M.; Haruka, O.; Sukegawa, S.; Tsujigiwa, H.; et al. The Role of Sonic Hedgehog Signaling in the Tumor Microenvironment of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5779. [Google Scholar] [CrossRef]
- Avery, J.T.; Zhang, R.; Boohaker, R.J. GLI1: A Therapeutic Target for Cancer. Front. Oncol. 2021, 11, 673154. [Google Scholar] [CrossRef]
- Lei, X.; Li, Z.; Huang, M.; Huang, L.; Huang, Y.; Lv, S.; Zhang, W.; Chen, Z.; Ke, Y.; Li, S.; et al. Gli1-mediated tumor cell-derived bFGF promotes tumor angiogenesis and pericyte coverage in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2024, 43, 83. [Google Scholar] [CrossRef]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.-W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef]
- Mirza, A.N.; McKellar, S.A.; Urman, N.M.; Brown, A.S.; Hollmig, T.; Aasi, S.Z.; Oro, A.E. LAP2 Proteins Chaperone GLI1 Movement between the Lamina and Chromatin to Regulate Transcription. Cell 2019, 176, 198–212.e115. [Google Scholar] [CrossRef]
- Tian, X.; Srinivasan, P.R.; Tajiknia, V.; Sanchez Sevilla Uruchurtu, A.F.; Seyhan, A.A.; Carneiro, B.A.; De La Cruz, A.; Pinho-Schwermann, M.; George, A.; Zhao, S.; et al. Targeting apoptotic pathways for cancer therapy. J. Clin. Investig. 2024, 134, e179570. [Google Scholar] [CrossRef]
- Lopez, A.; Reyna, D.E.; Gitego, N.; Kopp, F.; Zhou, H.; Miranda-Roman, M.A.; Nordstrøm, L.U.; Narayanagari, S.-R.; Chi, P.; Vilar, E.; et al. Co-targeting of BAX and BCL-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat. Commun. 2022, 13, 1199. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-e.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Zhu, J.; Deng, S.; Shi, H.; Xu, S.; Wu, H.; Zou, F. Targeting BAX ubiquitin-binding sites reveals that BAX activation is essential for its ubiquitin-dependent degradation. J. Cell. Biochem. 2020, 121, 2802–2810. [Google Scholar] [CrossRef] [PubMed]
- Cursi, S.; Rufini, A.; Stagni, V.; Condò, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Raab, M.; Matthess, Y.; Becker, S.; Knecht, R.; Strebhardt, K. pERK 1/2 inhibit Caspase-8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner. Mol. Oncol. 2014, 8, 232–249. [Google Scholar] [CrossRef]
- Nair, P.; Lu, M.; Petersen, S.; Ashkenazi, A. Chapter Five—Apoptosis Initiation Through the Cell-Extrinsic Pathway. In Methods in Enzymology; Ashkenazi, A., Yuan, J., Wells, J.A., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 544, pp. 99–128. [Google Scholar]
- Roberts, J.Z.; Crawford, N.; Longley, D.B. The role of Ubiquitination in Apoptosis and Necroptosis. Cell Death Differ. 2022, 29, 272–284. [Google Scholar] [CrossRef]
- Nam, Y.W.; Shin, J.-H.; Kim, S.; Hwang, C.H.; Lee, C.-S.; Hwang, G.; Kim, H.-R.; Roe, J.-S.; Song, J. EGFR inhibits TNF-α-mediated pathway by phosphorylating TNFR1 at tyrosine 360 and 401. Cell Death Differ. 2024, 31, 1318–1332. [Google Scholar] [CrossRef]
- Contadini, C.; Ferri, A.; Cirotti, C.; Stupack, D.; Barilà, D. Caspase-8 and Tyrosine Kinases: A Dangerous Liaison in Cancer. Cancers 2023, 15, 3271. [Google Scholar] [CrossRef]
- Liu, Z.; Ding, Y.; Ye, N.; Wild, C.; Chen, H.; Zhou, J. Direct Activation of Bax Protein for Cancer Therapy. Med. Res. Rev. 2016, 36, 313–341. [Google Scholar] [CrossRef]
- Yan, D.; Li, X.; Yang, Q.; Huang, Q.; Yao, L.; Zhang, P.; Sun, W.; Lin, S.; Dou, Q.P.; Liu, J.; et al. Regulation of Bax-dependent apoptosis by mitochondrial deubiquitinase USP30. Cell Death Discov. 2021, 7, 211. [Google Scholar] [CrossRef]
- Kale, J.; Kutuk, O.; Brito, G.C.; Andrews, T.S.; Leber, B.; Letai, A.; Andrews, D.W. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018, 19, e45235. [Google Scholar] [CrossRef]
- Benard, G.; Neutzner, A.; Peng, G.; Wang, C.; Livak, F.; Youle, R.J.; Karbowski, M. IBRDC2, an IBR-type E3 ubiquitin ligase, is a regulatory factor for Bax and apoptosis activation. EMBO J. 2010, 29, 1458–1471. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.T.; Stehlik, C.; Leonard, S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Ding, H.; Meng, X.W.; Lee, S.-H.; Schneider, P.A.; Kaufmann, S.H. Contribution of Bcl-2 Phosphorylation to Bak Binding and Drug Resistance. Cancer Res. 2013, 73, 6998–7008. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Tarcsafalvi, A.; Seng, N.; Hodeify, R.; Price, P.M. Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak. Cell Death Discov. 2016, 2, 15066. [Google Scholar] [CrossRef]
- Yoo, J.; Lee, S.; Jeong, E.-G.; Lee, S.; Yoo, N.J.; Lee, S.H.; Jeong, E.G. Expression of phosphorylated caspase-9 in gastric carcinomas. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 354–359. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Wei, W.; Qin, B.; Wen, W.; Zhang, B.; Luo, H.; Wang, Y.; Xu, H.; Xie, X.; Liu, S.; Jiang, X.; et al. FBXW7β loss-of-function enhances FASN-mediated lipogenesis and promotes colorectal cancer growth. Signal Transduct. Target. Ther. 2023, 8, 187. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Guo, D.; Meng, Y.; Jiang, X.; Lu, Z. Hexokinases in cancer and other pathologies. Cell Insight 2023, 2, 100077. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Ciccarone, F.; Ciriolo, M.R. Editorial: Hallmark of cancer: Sustained proliferative signalling. Front. Oncol. 2023, 13, 1328827. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Pardella, E.; Pranzini, E.; Raugei, G.; Paoli, P. Role of tyrosine phosphorylation in modulating cancer cell metabolism. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188442. [Google Scholar] [CrossRef] [PubMed]
- Yozgat, Y.; Karakoc, E.; Sahin, O.; Cimen, S.; Rabeh, W.M.; Aydin, M.S.; Mardinoglu, A.; Gursel, I.; Cakir, A.; Sensoy, O.; et al. Hexokinase 1b is a novel target for Non–small-cell lung cancer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lee, H.-J.; Li, C.-F.; Ruan, D.; He, J.; Montal, E.D.; Lorenz, S.; Girnun, G.D.; Chan, C.-H. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat. Commun. 2019, 10, 2625. [Google Scholar] [CrossRef]
- Li, H.; Lu, S.; Chen, Y.; Zheng, L.; Chen, L.; Ding, H.; Ding, J.; Lou, D.; Liu, F.; Zheng, B. AKT2 phosphorylation of hexokinase 2 at T473 promotes tumorigenesis and metastasis in colon cancer cells via NF-κB, HIF1α, MMP2, and MMP9 upregulation. Cell Signal. 2019, 58, 99–110. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Jiang, B.; Huang, L.; Ji, Z.; Li, X.; Zhou, H.; Han, A.; Chen, A.; Wu, Y.; et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat. Commun. 2017, 8, 13732. [Google Scholar] [CrossRef]
- Zhou, Z.; Zheng, X.; Zhao, J.; Yuan, A.; Lv, Z.; Shao, G.; Peng, B.; Dong, M.-Q.; Xu, Q.; Xu, X.; et al. ULK1-dependent phosphorylation of PKM2 antagonizes O-GlcNAcylation and regulates the Warburg effect in breast cancer. Oncogene 2024, 43, 1769–1778. [Google Scholar] [CrossRef]
- Vanauberg, D.; Schulz, C.; Lefebvre, T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: A promising target in anti-cancer therapies. Oncogenesis 2023, 12, 16. [Google Scholar] [CrossRef]
- Jin, Q.; Yuan, L.X.; Boulbes, D.; Baek, J.M.; Wang, Y.N.; Gomez-Cabello, D.; Hawke, D.H.; Yeung, S.C.; Lee, M.H.; Hortobagyi, G.N.; et al. Fatty acid synthase phosphorylation: A novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010, 12, R96. [Google Scholar] [CrossRef]
- Thongchot, S.; Aksonnam, K.; Thuwajit, P.; Yenchitsomanus, P.-T.; Thuwajit, C. Nucleolin-based targeting strategies in cancer treatment: Focus on cancer immunotherapy (Review). Int. J. Mol. Med. 2023, 52, 81. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.A.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Janic, A.; Abad, E.; Amelio, I. Decoding p53 tumor suppression: A crosstalk between genomic stability and epigenetic control? Cell Death Differ. 2024. [Google Scholar] [CrossRef] [PubMed]
- Feroz, W.; Sheikh, A.M.A. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Wen, J.; Wang, D. Deciphering the PTM codes of the tumor suppressor p53. J. Mol. Cell Biol. 2021, 13, 774–785. [Google Scholar] [CrossRef]
- Marvalim, C.; Datta, A.; Lee, S.C. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics 2023, 13, 1421–1442. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef]
- Lahusen, T.J.; Kim, S.-J.; Miao, K.; Huang, Z.; Xu, X.; Deng, C.-X. BRCA1 function in the intra-S checkpoint is activated by acetylation via a pCAF/SIRT1 axis. Oncogene 2018, 37, 2343–2350. [Google Scholar] [CrossRef]
- Densham, R.M.; Morris, J.R. The BRCA1 Ubiquitin ligase function sets a new trend for remodelling in DNA repair. Nucleus 2017, 8, 116–125. [Google Scholar] [CrossRef]
- Ouchi, T. BRCA1 phosphorylation: Biological consequences. Cancer Biol. Ther. 2006, 5, 470–475. [Google Scholar] [CrossRef]
- Salas-Lloret, D.; García-Rodríguez, N.; Soto-Hidalgo, E.; González-Vinceiro, L.; Espejo-Serrano, C.; Giebel, L.; Mateos-Martín, M.L.; de Ru, A.H.; van Veelen, P.A.; Huertas, P.; et al. BRCA1/BARD1 ubiquitinates PCNA in unperturbed conditions to promote continuous DNA synthesis. Nat. Commun. 2024, 15, 4292. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, F.; Liu, X.; Peng, B.; Xu, X.; Zhang, H.; Fu, L.; Zhu, W.-G.; Wang, B.; Pei, X.-H. GATA3 functions downstream of BRCA1 to promote DNA damage repair and suppress dedifferentiation in breast cancer. BMC Biol. 2024, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jin, J.; Yang, S.; Xu, W.; Meng, X.; Deng, H.; Zhan, J.; Gao, S.; Zhang, H. GATA3 acetylation at K119 by CBP inhibits cell migration and invasion in lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Izzo, F.; Mercogliano, F.; Venturutti, L.; Tkach, M.; Inurrigarro, G.; Schillaci, R.; Cerchietti, L.; Elizalde, P.V.; Proietti, C.J. Progesterone receptor activation downregulates GATA3 by transcriptional repression and increased protein turnover promoting breast tumor growth. Breast Cancer Res. 2014, 16, 491. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Pavitra, E.; Kancharla, J.; Gupta, V.K.; Prasad, K.; Sung, J.Y.; Kim, J.; Tej, M.B.; Choi, R.; Lee, J.-H.; Han, Y.-K.; et al. The role of NF-κB in breast cancer initiation, growth, metastasis, and resistance to chemotherapy. Biomed. Pharmacother. 2023, 163, 114822. [Google Scholar] [CrossRef]
- Devanaboyina, M.; Kaur, J.; Whiteley, E.; Lin, L.; Einloth, K.; Morand, S.; Stanbery, L.; Hamouda, D.; Nemunaitis, J. NF-κB Signaling in Tumor Pathways Focusing on Breast and Ovarian Cancer. Oncol. Rev. 2022, 16, 10568. [Google Scholar] [CrossRef]
- Al-Mutairi, M.S.; Habashy, H.O. Nuclear Factor-κB Clinical Significance in Breast Cancer: An Immunohistochemical Study. Med. Princ. Pract. 2023, 32, 33–39. [Google Scholar] [CrossRef]
- Abdrabou, A.M. The Yin and Yang of IκB Kinases in Cancer. Kinases Phosphatases 2024, 2, 9–27. [Google Scholar] [CrossRef]
- Sabaawy, H.E.; Ryan, B.M.; Khiabanian, H.; Pine, S.R. JAK/STAT of all trades: Linking inflammation with cancer development, tumor progression and therapy resistance. Carcinogenesis 2021, 42, 1411–1419. [Google Scholar] [CrossRef]
- To, S.Q.; Dmello, R.S.; Richards, A.K.; Ernst, M.; Chand, A.L. STAT3 Signaling in Breast Cancer: Multicellular Actions and Therapeutic Potential. Cancers 2022, 14, 429. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell. Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Bild, A.H.; Turkson, J.; Jove, R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. EMBO J. 2002, 21, 3255–3263. [Google Scholar] [CrossRef]
- Pérez-González, A.; Bévant, K.; Blanpain, C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 2023, 4, 1063–1082. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221.e3211. [Google Scholar] [CrossRef]
- Lin, W.-H.; Cooper, L.M.; Anastasiadis, P.Z. Cadherins and catenins in cancer: Connecting cancer pathways and tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1137013. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R. The SUMO guards for SNAIL. Oncotarget 2017, 8, 97701–97702. [Google Scholar] [CrossRef]
- Rodríguez-Alonso, A.; Casas-Pais, A.; Roca-Lema, D.; Graña, B.; Romay, G.; Figueroa, A. Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer. Cancers 2020, 12, 3093. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Won, J.-Y.; Kim, C.-H.; Kim, D.-E.; Yim, H. Roles of the Phosphorylation of Transcriptional Factors in Epithelial-Mesenchymal Transition. J. Oncol. 2019, 2019, 5810465. [Google Scholar] [CrossRef] [PubMed]
- Na, T.-Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.l.; Wang, S.-H.; Chan, P.-C.; Shen, M.-R.; Chen, H.-C. Phosphorylation of E-cadherin at threonine 790 by protein kinase Cδ reduces β-catenin binding and suppresses the function of E-cadherin. Oncotarget 2016, 7, 37260–37276. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Hua, F.; Hu, Z.-w. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef]
- You, H.; Li, Q.; Kong, D.; Liu, X.; Kong, F.; Zheng, K.; Tang, R. The interaction of canonical Wnt/β-catenin signaling with protein lysine acetylation. Cell. Mol. Biol. Lett. 2022, 27, 7. [Google Scholar] [CrossRef]
- Huang, H.-J.; Zhou, L.-L.; Fu, W.-J.; Zhang, C.-Y.; Jiang, H.; Du, J.; Hou, J. β-catenin SUMOylation is involved in the dysregulated proliferation of myeloma cells. Am. J. Cancer Res. 2015, 5, 309–320. [Google Scholar]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef]
- Muqbil, I.; Wu, J.; Aboukameel, A.; Mohammad, R.M.; Azmi, A.S. Snail nuclear transport: The gateways regulating epithelial-to-mesenchymal transition? Semin. Cancer Biol. 2014, 27, 39–45. [Google Scholar] [CrossRef]
- Sun, K.; Liao, S.; Yao, X.; Yao, F. USP30 promotes the progression of breast cancer by stabilising Snail. Cancer Gene Ther. 2024, 31, 472–483. [Google Scholar] [CrossRef]
- Gudey, S.K.; Sundar, R.; Mu, Y.; Wallenius, A.; Zang, G.; Bergh, A.; Heldin, C.-H.; Landström, M. TRAF6 Stimulates the Tumor-Promoting Effects of TGFβ Type I Receptor Through Polyubiquitination and Activation of Presenilin 1. Sci. Signal. 2014, 7, ra2. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xin, Y.; Xu, W.; Tian, X.; Wei, X.; Zhang, H. CBP-mediated Slug acetylation stabilizes Slug and promotes EMT and migration of breast cancer cells. Sci. China Life Sci. 2021, 64, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, Y.K.; Kim, H.; Lee, J.; Oh, M.J.; Kim, S.B.; Kim, M.; Kim, K.H.; Yoon, H.J.; Lee, M.-S.; et al. Snail acetylation by autophagy-derived acetyl-coenzyme A promotes invasion and metastasis of KRAS-LKB1 co-mutated lung cancer cells. Cancer Commun. 2022, 42, 716–749. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Gu, S.; Yu, Y.; Feng, Y.; Xiao, M.; Feng, X.-H. ZNF451 stabilizes TWIST2 through SUMOylation and promotes epithelial-mesenchymal transition. Am. J. Cancer Res. 2021, 11, 898–915. [Google Scholar]
- Costa, P.M.d.S.; Sales, S.L.A.; Pinheiro, D.P.; Pontes, L.Q.; Maranhão, S.S.A.; Pessoa, C.d.Ó.; Furtado, G.P.; Furtado, C.L.M. Epigenetic reprogramming in cancer: From diagnosis to treatment. Front. Cell Dev. Biol. 2023, 11, 1116805. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, M.; Wang, Y. The roles of histone modifications in tumorigenesis and associated inhibitors in cancer therapy. J. Natl. Cancer Cent. 2022, 2, 277–290. [Google Scholar] [CrossRef]
- Mravec, B. Nonmutational Epigenetic Reprogramming. In Neurobiology of Cancer: Role of the Nervous System in Cancer Etiopathogenesis, Treatment, and Prevention; Springer Nature: Cham, Switzerland, 2024; pp. 317–319. [Google Scholar]
- Zhang, J.; Zhao, Y.; Liang, R.; Zhou, X.; Wang, Z.; Yang, C.; Gao, L.; Zheng, Y.; Shao, H.; Su, Y.; et al. DNMT3A loss drives a HIF-1-dependent synthetic lethality to HDAC6 inhibition in non-small cell lung cancer. Acta Pharm. Sin. B 2024, 14, 5219–5234. [Google Scholar] [CrossRef]
- Seo, J.; Li, L.; Small, D. Dissociation of the DNMT3A-HDAC1 Repressor Complex Induces PD-L1 Expression. Blood 2019, 134, 3759. [Google Scholar] [CrossRef]
- Ling, Y.; Sankpal, U.T.; Robertson, A.K.; McNally, J.G.; Karpova, T.; Robertson, K.D. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res. 2004, 32, 598–610. [Google Scholar] [CrossRef]
- Dupas, T.; Lauzier, B.; McGraw, S. O-GlcNAcylation: The sweet side of epigenetics. Epigenetics Chromatin 2023, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. N ε-Formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 2007, 36, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guan, J.; Huang, Z.; Hu, X.; Zheng, X. RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J. Cell Sci. 2014, 127, 2238–2248. [Google Scholar] [CrossRef] [PubMed]
- Neja, S.; Dashwood, W.M.; Dashwood, R.H.; Rajendran, P. Histone Acyl Code in Precision Oncology: Mechanistic Insights from Dietary and Metabolic Factors. Nutrients 2024, 16, 396. [Google Scholar] [CrossRef]
- Ntorla, A.; Burgoyne, J.R. The Regulation and Function of Histone Crotonylation. Front. Cell Dev. Biol. 2021, 9, 624914. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, K.; Wang, Y.; Gu, Y. Comprehensive review of histone lactylation: Structure, function, and therapeutic targets. Biochem. Pharmacol. 2024, 225, 116331. [Google Scholar] [CrossRef]
- Kebede, A.F.; Nieborak, A.; Shahidian, L.Z.; Le Gras, S.; Richter, F.; Gómez, D.A.; Baltissen, M.P.; Meszaros, G.; Magliarelli, H.d.F.; Taudt, A.; et al. Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol. 2017, 24, 1048–1056. [Google Scholar] [CrossRef]
- Liu, J.; Shangguan, Y.; Tang, D.; Dai, Y. Histone succinylation and its function on the nucleosome. J. Cell. Mol. Med. 2021, 25, 7101–7109. [Google Scholar] [CrossRef]
- Kuroishi, T.; Rios-Avila, L.; Pestinger, V.; Wijeratne, S.S.K.; Zempleni, J. Biotinylation is a natural, albeit rare, modification of human histones. Mol. Genet. Metab. 2011, 104, 537–545. [Google Scholar] [CrossRef]
- Kothapalli, N.; Camporeale, G.; Kueh, A.; Chew, Y.C.; Oommen, A.M.; Griffin, J.B.; Zempleni, J. Biological functions of biotinylated histones. J. Nutr. Biochem. 2005, 16, 446–448. [Google Scholar] [CrossRef]
- Zentout, S.; Imburchia, V.; Chapuis, C.; Duma, L.; Schützenhofer, K.; Prokhorova, E.; Ahel, I.; Smith, R.; Huet, S. Histone ADP-ribosylation promotes resistance to PARP inhibitors by facilitating PARP1 release from DNA lesions. Proc. Natl. Acad. Sci. USA 2024, 121, e2322689121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020, 5, 143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, S.Y.; Choi, J.-H.; Woo, H.G.; Xhemalce, B.; Miller, K.M. PCAF-Mediated Histone Acetylation Promotes Replication Fork Degradation by MRE11 and EXO1 in BRCA-Deficient Cells. Mol. Cell 2020, 80, 327–344.e328. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Lythgoe, M.P.; Mullish, B.H.; Frampton, A.E.; Krell, J. Polymorphic microbes: A new emerging hallmark of cancer. Trends Microbiol. 2022, 30, 1131–1134. [Google Scholar] [CrossRef]
- Mafra, D.; Ribeiro, M.; Fonseca, L.; Regis, B.; Cardozo, L.F.M.F.; Fragoso dos Santos, H.; Emiliano de Jesus, H.; Schultz, J.; Shiels, P.G.; Stenvinkel, P.; et al. Archaea from the gut microbiota of humans: Could be linked to chronic diseases? Anaerobe 2022, 77, 102629. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Papakonstantinou, A.; Nuciforo, P.; Borrell, M.; Zamora, E.; Pimentel, I.; Saura, C.; Oliveira, M. The conundrum of breast cancer and microbiome—A comprehensive review of the current evidence. Cancer Treat. Rev. 2022, 111, 102470. [Google Scholar] [CrossRef]
- Mendes, I.; Vale, N. How Can the Microbiome Induce Carcinogenesis and Modulate Drug Resistance in Cancer Therapy? Int. J. Mol. Sci. 2023, 24, 11855. [Google Scholar] [CrossRef]
- Zhou, X.; Kandalai, S.; Hossain, F.; Zheng, Q. Tumor microbiome metabolism: A game changer in cancer development and therapy. Front. Oncol. 2022, 12, 933407. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Islam, M.R.; Shohag, S.; Ahasan, M.T.; Sarkar, N.; Khan, H.; Hasan, A.M.; Cavalu, S.; Rauf, A. Microbiome in cancer: Role in carcinogenesis and impact in therapeutic strategies. Biomed. Pharmacother. 2022, 149, 112898. [Google Scholar] [CrossRef] [PubMed]
- Cao, H. Bacterial endotoxin lipopolysaccharides regulate gene expression in human colon cancer cells. BMC Res. Notes 2023, 16, 216. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kundu, P.; Seow, S.W.; de Matos, C.T.; Aronsson, L.; Chin, K.C.; Kärre, K.; Pettersson, S.; Greicius, G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APC Min/+ mice. Carcinogenesis 2012, 33, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, C.; Zuo, D.; Zhang, T.; Yin, F.; Zhou, Z.; Wang, H.; Xu, J.; Mao, M.; Wang, G.; et al. Attenuation of STAT3 Phosphorylation Promotes Apoptosis and Chemosensitivity in Human Osteosarcoma Induced by Raddeanin A. Int. J. Biol. Sci. 2019, 15, 668–679. [Google Scholar] [CrossRef]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Helicobacter pylori and gastric cancer: A critical approach to who really needs eradication. Explor. Dig. Dis. 2024, 3, 107–142. [Google Scholar] [CrossRef]
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA Phosphorylation in Helicobacter pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect. Immun. 2016, 84, 2671–2680. [Google Scholar] [CrossRef]
- Piao, J.-Y.; Kim, S.-J.; Kim, D.-H.; Park, J.H.; Park, S.-A.; Han, H.-j.; Na, H.-K.; Yoon, K.; Lee, H.-N.; Kim, N.; et al. Helicobacter pylori infection induces STAT3 phosphorylation on Ser727 and autophagy in human gastric epithelial cells and mouse stomach. Sci. Rep. 2020, 10, 15711. [Google Scholar] [CrossRef]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial Localized Stat3 Promotes Breast Cancer Growth via Phosphorylation of Serine 727 *. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Zheng, Z.-M. Viral Oncogenes, Noncoding RNAs, and RNA Splicing in Human Tumor Viruses. Int. J. Biol. Sci. 2010, 6, 730–755. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Malone, M.; Maeyama, A.; Ogden, N.; Perry, K.N.; Kramer, A.; Bates, C.; Marble, C.; Orlando, R.; Rausch, A.; Smeraldi, C.; et al. The effect of phosphorylation efficiency on the oncogenic properties of the protein E7 from high-risk HPV. Virus Res. 2024, 348, 199446. [Google Scholar] [CrossRef] [PubMed]
- Billimoria, R.; Bhatt, P. Senescence in cancer: Advances in detection and treatment modalities. Biochem. Pharmacol. 2023, 215, 115739. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, S.; Cheng, A.; Colmegna, I.; Rodier, F. Quantifying Senescence-Associated Phenotypes in Primary Multipotent Mesenchymal Stromal Cell Cultures. In Stem Cells and Aging: Methods and Protocols; Turksen, K., Ed.; Springer: New York, NY, USA, 2019; pp. 93–105. [Google Scholar]
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, E.A.; Wallis, R.; Mossa, F.; Bishop, C.L. The paradox of senescent-marker positive cancer cells: Challenges and opportunities. npj Aging 2024, 10, 41. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, W.; Chen, Y.; Huang, Y.; Zhang, J.; He, S.; Tan, Y.; Qiang, F.; Li, A.; Røe, O.D.; et al. The opposite prognostic significance of nuclear and cytoplasmic p21 expression in resectable gastric cancer patients. J. Gastroenterol. 2014, 49, 1441–1452. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Vincent, A.J.; Ren, S.; Harris, L.G.; Devine, D.J.; Samant, R.S.; Fodstad, O.; Shevde, L.A. Cytoplasmic translocation of p21 mediates NUPR1-induced chemoresistance. FEBS Lett. 2012, 586, 3429–3434. [Google Scholar] [CrossRef]
- Xia, W.; Chen, J.-S.; Zhou, X.; Sun, P.-R.; Lee, D.-F.; Liao, Y.; Zhou, B.P.; Hung, M.-C. Phosphorylation/Cytoplasmic Localization of p21Cip1/WAF1 Is Associated with HER2/neu Overexpression and Provides a Novel Combination Predictor for Poor Prognosis in Breast Cancer Patients. Clin. Cancer Res. 2004, 10, 3815–3824. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTMs of Proteins | Definition | Roles in Cancer | References |
|---|---|---|---|
| phosphorylation | attachment of a phosphate group to specific amino acid residues of a protein | PPIs, cell growth, proliferation, division, differentiation, apoptosis, cell signaling, angiogenesis, metastasis | [31,32,33] |
| methylation | transfer of active methyl group to target histone or non-histone proteins | protein activity, localization, signaling, regulator of MAPK, WNT, JAK-STAT, Hippo, p53, NF-kB signaling | [34] |
| ubiquitination/deubiquitination | tagging of specifically targeted proteins with ubiquitin for degradation, translocation, or activation/reverse process of ubiquitination | proteostasis (protein stability/activity/degradation/turnover), DDR, immune response, cancer cell stemness, proliferation via cell cycle progression, all cancer hallmarks (evading growth suppressors, evasion of apoptosis, reprogramming energy metabolism, unlocking phenotypic plasticity, polymorphic microbiomes, immune evasion, resisting cell death, tumor-promoting inflammation, senescent cells) | [35,36,37,38] |
| SUMOylation | covalent attachment of a SUMO molecule to a substrate protein | chromatin organization, DDR, transcription, protein trafficking, signal transduction, TME formation and reprogramming, carcinogenesis, immune responses, cell cycle progression, apoptosis, metastasis | [39,40] |
| NEDDylation | attachment of NEDD8 to a lysine residue of a protein substrate (cullins and non-cullins proteins) | tumor progression (proliferation), increased malignancy, cancer cell behavior, regulation of protein degradation, angiogenesis, modulation of ECM, regulation of T-cell functionality | [28,41,42] |
| glycosylation | addition of individual carbohydrates or whole oligosaccharides (N-glycans, O-glycans, and proteoglycans) to corresponding protein or lipid | protein folding, secretion, cell adhesion, intra- and intercellular trafficking, cancer progression, cancer cell stemness, EMT, avoiding immune destruction | [11,43] |
| O-linked-β-N-Acetylglucosaminylation (O-GlcNAcylation) | addition of N-acetylglucosamine (GlcNAc) onto the hydroxyl groups of the Ser or Thr residues of proteins | development, maturation, and functions of immune cells, signal transduction, transcription, cell division, metabolism, and cytoskeletal regulation | [44] |
| acylation | transfer of acyl groups from acyl-CoA donors to sidechain of lysine (glycine, cysteine, serine, and others) residues of proteins | protein stability, subcellular localization, enzyme activity, transcriptional activity, PPIs, protein-DNA interactions | [19,45] |
| acetylation (Kac) | attachment of acetyl group from acetyl-coenzyme A to a specific site on a polypeptide chain | gene expression, cell cycle progress, DDR, PPIs, protein-DNA interactions, cell proliferation | [18] |
| propionylation (Kpr) | addition of propionyl groups to specific amino acid residues in a protein | modification of protein structure and functions, gene regulation, metabolic pathway, cellular signaling networks | [46] |
| succinylation (ksucc) | attachment of a succinyl group to a lysine residue | tumorigenesis, transcriptional regulation of genes, energy metabolism | [22] |
| crotonylation (Kcr) | transfers of crotonyl group onto lysine residues using crotonyl-CoA as substrate under the action of crotonyltransferase | carcinogenesis, tumor progression | [21,47] |
| glutarylation (Kglu) | addition a five-carbon glutaryl group to lysine residue non-enzymatically driven by glutaryl-CoA | regulation of mitochondrial proteins and metabolic enzymes, regulation of gene expression | [19] |
| β-hydroxybutyrylation (Kbhb) | addition of β-hydroxybutyrate to lysine residue | attenuates ALDOB activity in ketogenic diet, inhibits mTOR, glycolysis, and suppresses cancer cell proliferation | [23,24] |
| 2-hydroxybutyrylation (Khib) | addition of butanoate to lysine residues | chromatin structure, gene transcription, protein subcellular localization, PPIs, signal transduction, glucose metabolism, amino acid synthesis, cell proliferation | [48,49,50] |
| malonylation (Kmal) | attachment of a malonyl group to a lysine residue | protein activity, localization, PPIs | [20] |
| lactylation (Kla) | covalent attachment of lactic acid moieties to protein lysine residues | gene transcription/expression regulation, cancer progression | [51,52] |
| benzonylation (Kbz) | introduction of benzoyl group by replacement of H- attached to O or N or to aromatic nucleus, stimulated by sodium benzoate through benzoyl-CoA generation | gene transcription regulation | [53] |
| S-palmitoylation/S-acylation | reversible attachment of fatty acids onto cysteine residue | protein localization, membrane affinity, stability, accumulation, secretion, and function, PPIs; cell proliferation | [10,54] |
| S-nitrosylation | addition of a nitrosyl group to the reactive thiol group of cysteine to form S-nitrosothiol | protein stability and turnover, steroid synthesis, transcription regulation, DDR, cellular growth, apoptosis, and redox regulation | [29] |
| citrullination/arginine deimination | converts arginine residues in proteins to citrulline; human citrullinome include VIM, ACTs, COL, FN, CKs, TUBs, and histones | protein folding, PPIs, regulation of apoptosis and differentiation, promotion of EMT, and metastasis | [8,55,56,57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, P.S.; Arshad, A.; Gogu, M.-R.; Waterman, N.; Flack, R.; Dunn, K.; Darie, C.C.; Neagu, A.-N. Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer. Life 2025, 15, 126. https://doi.org/10.3390/life15010126
Bruno PS, Arshad A, Gogu M-R, Waterman N, Flack R, Dunn K, Darie CC, Neagu A-N. Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer. Life. 2025; 15(1):126. https://doi.org/10.3390/life15010126
Chicago/Turabian StyleBruno, Pathea Shawnae, Aneeta Arshad, Maria-Raluca Gogu, Natalie Waterman, Rylie Flack, Kimberly Dunn, Costel C. Darie, and Anca-Narcisa Neagu. 2025. "Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer" Life 15, no. 1: 126. https://doi.org/10.3390/life15010126
APA StyleBruno, P. S., Arshad, A., Gogu, M.-R., Waterman, N., Flack, R., Dunn, K., Darie, C. C., & Neagu, A.-N. (2025). Post-Translational Modifications of Proteins Orchestrate All Hallmarks of Cancer. Life, 15(1), 126. https://doi.org/10.3390/life15010126