
Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review


Abstract
:1. Introduction
2. Results
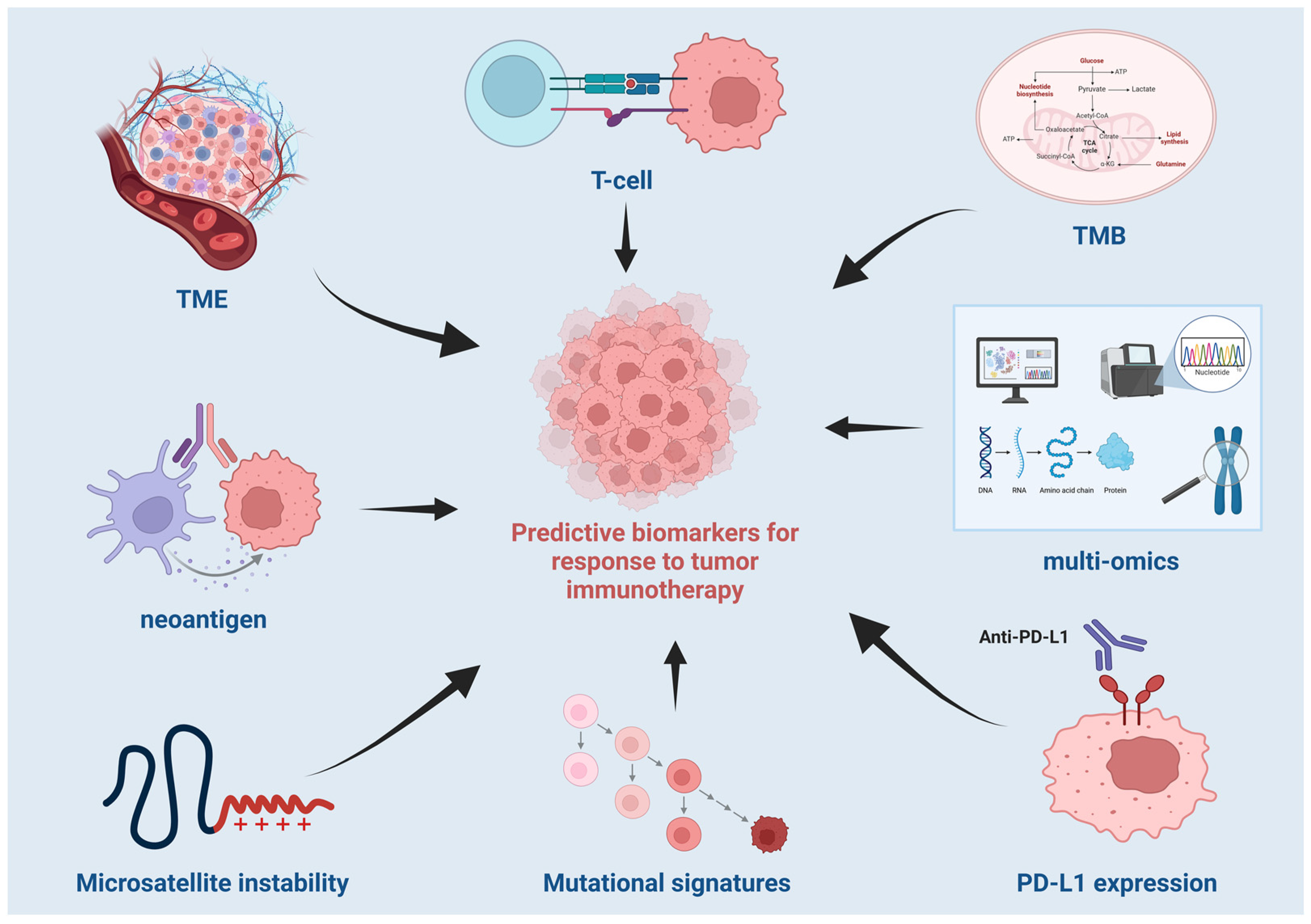
2.1. Predictive Biomarkers for Response to Tumor Immunotherapy
2.1.1. Sequence-Based Biomarkers
Tumor Mutation Burden
Neoantigen
Microsatellite Instability
Mutational Signatures
2.1.2. Biomarkers Based on Gene Expression Profiles
2.1.3. Biomarkers Based on Epigenomic Profiles
DNA Methylation Profiles
Histone Modification Profiles
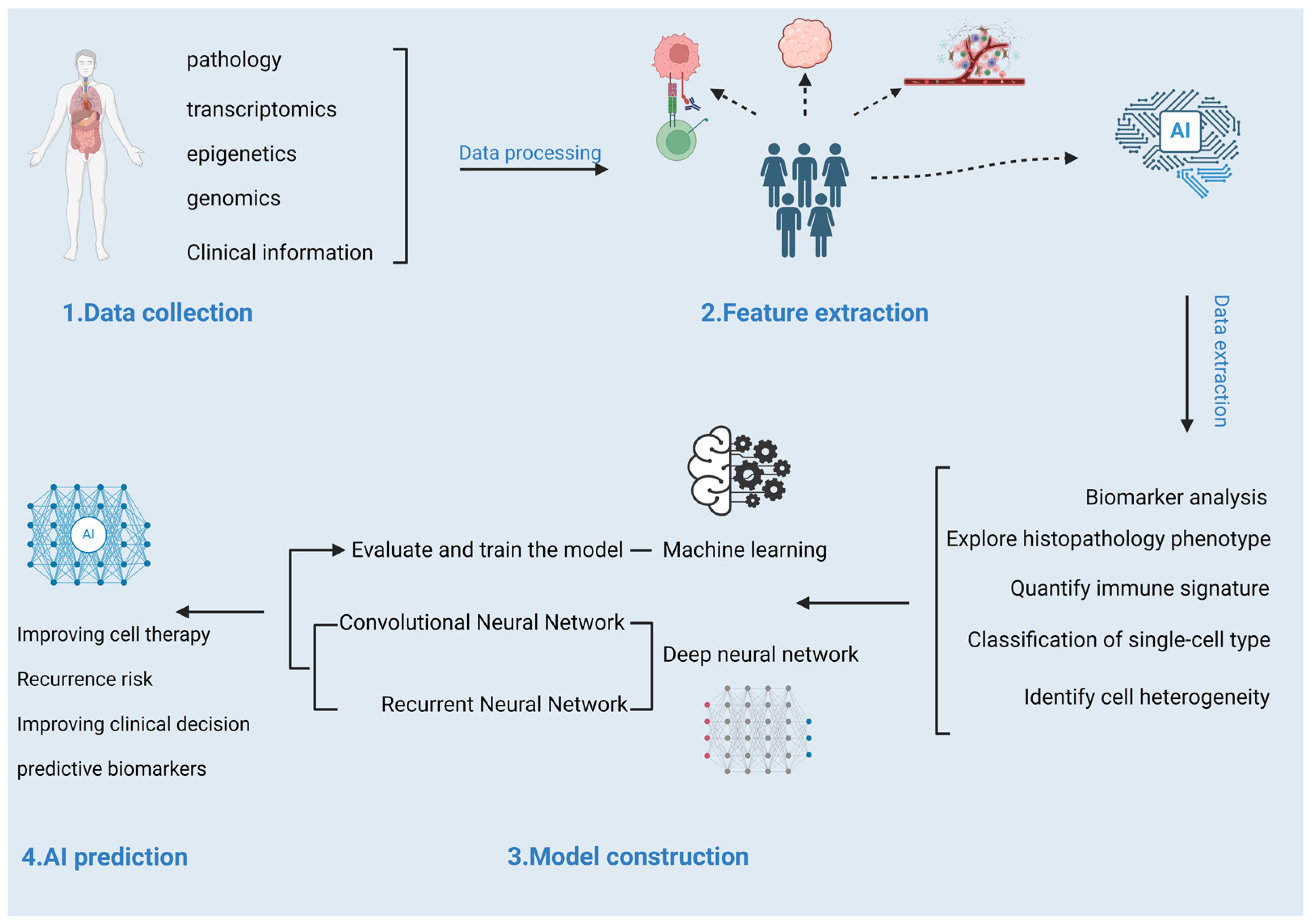
2.2. Cohorts and Datasets for Studying Tumor Immunotherapy Responses
2.2.1. Designing Cohorts for Studying the Tumor Immunotherapy Responses
2.2.2. Application of Public Cancer Data Sources in Developing the Prediction Models for Tumor Immunotherapy Responses
2.2.3. Published Clinical Cohorts on Cancer Immunotherapy
2.3. Prediction Models for Response to Tumor Immunotherapy
2.3.1. Machine Learning-Based Prediction Models
2.3.2. Deep Neural Network-Based Prediction Models
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kong, X.; Zhang, J.; Chen, S.; Wang, X.; Xi, Q.; Shen, H.; Zhang, R. Immune checkpoint inhibitors: Breakthroughs in cancer treatment. Cancer Biol. Med. 2024, 21, 451–472. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.S.; Lim, A.M.; Angel, C.; Young, R.J.; Pizzolla, A.; Archer, S.; Solomon, B.J.; Thai, A.A.; Lewin, J.; Rischin, D. A Retrospective Review and Comprehensive Tumour Profiling of Advanced Non-Melanomatous Cutaneous Spindle Cell Neoplasms Treated with Immune-Checkpoint Inhibitors. Cancers 2024, 16, 1452. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Liu, X.; Ma, T.; Lv, D.; Sun, G. Predictive value of tumor mutational burden for immunotherapy in non-small cell lung cancer: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0263629. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, J.P.; Sánchez-Canteli, M.; Otero-Rosales, M.; Martínez-Camblor, P.; Hermida-Prado, F.; García-Pedrero, J.M. Tumor mutational burden predictability in head and neck squamous cell carcinoma patients treated with immunotherapy: Systematic review and meta-analysis. J. Transl. Med. 2024, 22, 135. [Google Scholar] [CrossRef]
- Garon Edward, B.; Rizvi Naiyer, A.; Hui, R.; Leighl, N.; Balmanoukian Ani, S.; Eder Joseph, P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Nghiem Paul, T.; Bhatia, S.; Lipson Evan, J.; Kudchadkar Ragini, R.; Miller Natalie, J.; Annamalai, L.; Berry, S.; Chartash Elliot, K.; Daud, A.; Fling Steven, P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Pan, Y.; Zou, S. Response to PD-1 inhibitor in SMARCB1-deficient undifferentiated rectal carcinoma with low TMB, proficient MMR and BRAF V600E mutation: A case report and literature review. Diagn. Pathol. 2024, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Sisoudiya, S.D.; Houle, A.A.; Fernando, T.; Wilson, T.R.; Schutzman, J.L.; Lee, J.; Schrock, A.; Sokol, E.S.; Sivakumar, S.; Shi, Z.; et al. Ancestry-associated co-alteration landscape of KRAS and EGFR-altered non-squamous NSCLC. npj Precis. Oncol. 2024, 8, 153. [Google Scholar] [CrossRef]
- Li, S.; Li, L.; Pan, T.; Li, X.; Tong, Y.; Jin, Y. Prognostic value of TIGIT in East Asian patients with solid cancers: A systematic review, meta-analysis and pancancer analysis. Front. Immunol. 2022, 13, 977016. [Google Scholar] [CrossRef]
- Nguyen, K.; Fama, K.; Mercado, G.; Myat, Y.; Thein, K. Histology Agnostic Drug Development: An Updated Review. Cancers 2024, 16, 3642. [Google Scholar] [CrossRef] [PubMed]
- Ozdogan, M.; Papadopoulou, E.; Metaxa-Mariatou, V.; Kapetsis, G.; Meintani, A.; Florou-Chatzigiannidou, C.; Yildiz, A.; Cakir, M.O.; Kirca, O.; Nasioulas, G. Case report: Immunotherapy guided by molecular profiling of tumors: Illustrative cases and literature review. Front. Med. 2024, 11, 1403056. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Chen, M.Y.; Goedegebuure, S.P.; Gillanders, W.E. Challenges targeting cancer neoantigens in 2021: A systematic literature review. Expert Rev. Vaccines 2021, 20, 827–837. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Vazquez, M.; Finotello, F.; Lepore, R.; Porta, E.; Hundal, J.; Amengual-Rigo, P.; Ng, C.K.Y.; Valencia, A.; Carrillo, J.; et al. Neoantigen prediction and computational perspectives towards clinical benefit: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Xie, H.; Lv, M.; Yang, Q.; Shuang, Z.; Gao, F.; Li, S.; Zhu, L.; Wang, W. The landscape of objective response rate of anti-PD-1/L1 monotherapy across 31 types of cancer: A system review and novel biomarker investigating. Cancer Immunol. Immunother. 2023, 72, 2483–2498. [Google Scholar] [CrossRef]
- Jiang, J.; Ding, Y.; Wu, M.; Chen, Y.; Lyu, X.; Lu, J.; Wang, H.; Teng, L. Integrated genomic analysis identifies a genetic mutation model predicting response to immune checkpoint inhibitors in melanoma. Cancer Med. 2020, 9, 8498–8518. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, M.; Ghidini, A.; Tomasello, G. Outcomes Following Immune Checkpoint Inhibitor Treatment of Patients with Microsatellite Instability-High Cancers: A Systematic Review and Meta-analysis. JAMA Oncol. 2020, 6, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e514. [Google Scholar] [CrossRef]
- Khan, M.; Li, X.; Yan, M.; Li, Z.; Yang, H.; Liao, G. Efficacy and Safety of Actively Personalized Neoantigen Vaccination in the Management of Newly Diagnosed Glioblastoma: A Systematic Review. Int. J. Gen. Med. 2021, 14, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Cunningham, D.; Metges, J.-P.; Van Cutsem, E.; Wainberg, Z.; Elboudwarej, E.; Lin, K.-W.; Turner, S.; Zavodovskaya, M.; Inzunza, D.; et al. Randomized, open-label, phase 2 study of andecaliximab plus nivolumab versus nivolumab alone in advanced gastric cancer identifies biomarkers associated with survival. J. Immunother. Cancer 2021, 9, e003580. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.M.; Nankivell, M.G.; Morano, F.; et al. Individual Patient Data Meta-Analysis of the Value of Microsatellite Instability As a Biomarker in Gastric Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef]
- Yoon, H.H.; Jin, Z.; Kour, O.; Kankeu Fonkoua, L.A.; Shitara, K.; Gibson, M.K.; Prokop, L.J.; Moehler, M.; Kang, Y.K.; Shi, Q.; et al. Association of PD-L1 Expression and Other Variables with Benefit From Immune Checkpoint Inhibition in Advanced Gastroesophageal Cancer: Systematic Review and Meta-analysis of 17 Phase 3 Randomized Clinical Trials. JAMA Oncol. 2022, 8, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Bogani, G.; Monk, B.J.; Powell, M.A.; Westin, S.N.; Slomovitz, B.; Moore, K.N.; Eskander, R.N.; Raspagliesi, F.; Barretina-Ginesta, M.P.; Colombo, N.; et al. Adding immunotherapy to first-line treatment of advanced and metastatic endometrial cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2024, 35, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Jiang, R.; Philip, P.A.; Diab, M.; Behera, M.; Wu, C.; Alese, O.; Shaib, W.L.; Gaines, T.M.; Balch, G.G.; et al. High-Risk Features Are Prognostic in dMMR/MSI-H Stage II Colon Cancer. Front. Oncol. 2021, 11, 755113. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, X.Q.; Zhao, G.Y.; Wang, F.J.; Liu, X. Meta-analysis of neoadjuvant immunotherapy for non-metastatic colorectal cancer. Front. Immunol. 2023, 14, 1044353. [Google Scholar] [CrossRef]
- Mitric, C.; Salman, L.; Abrahamyan, L.; Kim, S.R.; Pechlivanoglou, P.; Chan, K.K.W.; Gien, L.T.; Ferguson, S.E. Mismatch-repair deficiency, microsatellite instability, and lynch syndrome in ovarian cancer: A systematic review and meta-analysis. Gynecol. Oncol. 2023, 170, 133–142. [Google Scholar] [CrossRef]
- Formica, V.; Sera, F.; Cremolini, C.; Riondino, S.; Morelli, C.; Arkenau, H.T.; Roselli, M. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 517–527. [Google Scholar] [CrossRef]
- Quaas, A.; Biesma, H.D.; Wagner, A.D.; Verheij, M.; van Berge Henegouwen, M.I.; Schoemig-Markiefka, B.; Pamuk, A.; Zander, T.; Siemanowski, J.; Sikorska, K.; et al. Microsatellite instability and sex differences in resectable gastric cancer—A pooled analysis of three European cohorts. Eur. J. Cancer 2022, 173, 95–104. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, E.; Reynolds, I.S.; McNamara, D.A.; Prehn, J.H.M.; Burke, J.P. Microsatellite instability and response to neoadjuvant chemoradiotherapy in rectal cancer: A systematic review and meta-analysis. Surg. Oncol. 2020, 34, 57–62. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.; Degasperi, A.; Zou, X.; Momen, S.; Nik-Zainal, S. Mutational signatures: Emerging concepts, caveats and clinical applica tions. Nat. Rev. Cancer. 2021, 21, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Wang, Y.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and Their Stimulated Genes in the Tumor Microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, T.; Jiang, R.; Yang, X.; Guo, H.; Yang, R. Targeting MHC-I molecules for cancer: Function, mechanism, and therapeutic prospects. Mol. Cancer 2023, 22, 194. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Xu, R.; Ma, F.; Yang, N.; Li, Y.; Sun, X.; Jin, P.; Kang, W.; Jia, L.; Xiong, J.; et al. scRNA-seq of gastric tumor shows complex intercellular interaction with an alternative T cell exhaustion trajectory. Nat. Commun. 2022, 13, 4943. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
- Kovács, S.A.; Fekete, J.T.; Győrffy, B. Predictive biomarkers of immunotherapy response with pharmacological applications in solid tumors. Acta Pharmacol. Sin. 2023, 44, 1879–1889. [Google Scholar] [CrossRef]
- Verma, M. The Role of Epigenomics in the Study of Cancer Biomarkers and in the Development of Diagnostic Tools. In Advances in Cancer Biomarkers: From Biochemistry to Clinic for a Critical Revision; Scatena, R., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 59–80. [Google Scholar]
- Tulsyan, S.; Aftab, M.; Sisodiya, S.; Khan, A.; Chikara, A.; Tanwar, P.; Hussain, S. Molecular basis of epigenetic regulation in cancer diagnosis and treatment. Front. Genet. 2022, 13, 885635. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Duran-Ferrer, M.; Martín-Subero, J.I. Epigenomic Characterization of Lymphoid Neoplasms. Annu. Rev. Pathol. Mech. Dis. 2024, 19, 371–396. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.; Op de Beeck, K.; Fransen, E.; Peeters, M.; Van Camp, G. Genome-wide DNA methylation profiling and identification of potential pan-cancer and tumor-specific biomarkers. Mol. Oncol. 2022, 16, 2432–2447. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-W.; Hong, M.H.; Ha, S.-J.; Kim, Y.-J.; Cho, B.C.; Lee, I.; Kim, H.R. Genome-wide identification of differentially methylated promoters and enhancers associated with response to anti-PD-1 therapy in non-small cell lung cancer. Exp. Mol. Med. 2020, 52, 1550–1563. [Google Scholar] [CrossRef]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: A multicentre, retrospective analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef]
- Xu, B.; Lu, M.; Yan, L.; Ge, M.; Ren, Y.; Wang, R.; Shu, Y.; Hou, L.; Guo, H. A Pan-Cancer Analysis of Predictive Methylation Signatures of Response to Cancer Immunotherapy. Front. Immunol. 2021, 12, 796647. [Google Scholar] [CrossRef]
- Lacoursiere, R.E.; Hadi, D.; Shaw, G.S. Acetylation, Phosphorylation, Ubiquitination (Oh My!): Following Post-Translational Modifications on the Ubiquitin Road. Biomolecules 2022, 12, 467. [Google Scholar] [CrossRef]
- Yang, X.; Hu, B.; Hou, Y.; Qiao, Y.; Wang, R.; Chen, Y.; Qian, Y.; Feng, S.; Chen, J.; Liu, C.; et al. Silencing of developmental genes by H3K27me3 and DNA methylation reflects the discrepant plasticity of embryonic and extraembryonic lineages. Cell Res. 2018, 28, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shi, J.; Li, W.; Tanaka, K.; Allton, K.L.; Richardson, D.; Li, J.; Franco, H.L.; Nagari, A.; Malladi, V.S.; et al. Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genom. 2018, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, L.; Liu, Y.; Li, S. Sirtuin SIRT6 suppresses cell proliferation through inhibition of Twist1 expression in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4774–4781. [Google Scholar] [PubMed]
- Liu, Y.-X.; Li, Q.-Z.; Cao, Y.-N.; Zhang, L.-Q. Identification of key genes and important histone modifications in hepatocellular carcinoma. Comput. Struct. Biotechnol. J. 2020, 18, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Yu, Z.; Surineni, G.; Deng, B.; Zhang, M.; Li, C.; Sun, Z.; Pan, W.; Liu, Y.; Liu, S.; et al. Discovery of novel benzohydroxamate-based histone deacetylase 6 (HDAC6) inhibitors with the ability to potentiate anti-PD-L1 immunotherapy in melanoma. J. Enzym. Inhib. Med. Chem. 2023, 38, 2201408. [Google Scholar] [CrossRef]
- Mouti, M.A.; Deng, S.; Pook, M.; Malzahn, J.; Rendek, A.; Militi, S.; Nibhani, R.; Soonawalla, Z.; Oppermann, U.; Hwang, C.-i.; et al. KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics. Nat. Commun. 2023, 14, 5685. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Ye, M.; Jiang, M.; Chen, X.; Song, G.; Ji, H.; Wang, Z.-w.; Zhu, X. Methylation of BRD4 by PRMT1 regulates BRD4 phosphorylation and promotes ovarian cancer invasion. Cell Death Dis. 2023, 14, 624. [Google Scholar] [CrossRef]
- Yin, G.; Liu, X.; Yu, X.; Tan, S.; Liu, F. Analysis of ICIs alone or in combination rechallenged outcomes after progression from first-line ICIs plus chemotherapy in patients with advanced non-small cell lung cancer. Sci. Rep. 2025, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. Współczesna Onkol. 2015, 2015, 68–77. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination stra tegies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nie, J.; Li, X.; Fan, T.; Deng, X.; Liang, D.; Song, G. Identification of Immune-Related Prognostic Biomarkers Associated with HPV-Positive Head and Neck Squamous Cell Carcinoma. J. Immunol. Res. 2021, 2021, 6661625. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson Andrew, G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Ferris Robert, L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes Everett, E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo Hope, S.; Schneeweiss, A.; Barrios Carlos, H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long Georgina, V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel Jessica, C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Vathiotis, I.A.; Salichos, L.; Martinez-Morilla, S.; Gavrielatou, N.; Aung, T.N.; Shafi, S.; Wong, P.F.; Jessel, S.; Kluger, H.M.; Syrigos, K.N.; et al. Baseline gene expression profiling determines long-term benefit to programmed cell death protein 1 axis blockade. npj Precis. Oncol. 2022, 6, 92. [Google Scholar] [CrossRef]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.-M.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Esteller, M.; Lee, S.-H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, Y.; Zhu, X.; He, Y.; Wu, Y.; Ying, T.; Xie, Z. Artificial intelligence in cancer immunotherapy: Applications in neoantigen recognition, antibody design and immunotherapy response prediction. Semin. Cancer Biol. 2023, 91, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Liao, H.; Yun, H.; Lin, N.; Li, S.; Xiang, Y.; Ma, X. Artificial intelligence-based prediction of clinical outcome in immunotherapy and targeted therapy of lung cancer. Semin. Cancer Biol. 2022, 86, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Painuli, D.; Bhardwaj, S.; Köse, U. Recent advancement in cancer diagnosis using machine learning and deep learning techniques: A comprehensive review. Comput. Biol. Med. 2022, 146, 105580. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef]
- Ma, B.; Meng, F.; Yan, G.; Yan, H.; Chai, B.; Song, F. Diagnostic classification of cancers using extreme gradient boosting algorithm and multi-omics data. Comput. Biol. Med. 2020, 121, 103761. [Google Scholar] [CrossRef]
- Statnikov, A.; Wang, L.; Aliferis, C.F. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinform. 2008, 9, 319. [Google Scholar] [CrossRef]
- Ning, B.; Chi, J.; Meng, Q.; Jia, B. Accurate prediction of colorectal cancer diagnosis using machine learning based on immunohistochemistry pathological images. Sci. Rep. 2024, 14, 29882. [Google Scholar] [CrossRef] [PubMed]
- Moon, I.; LoPiccolo, J.; Baca, S.C.; Sholl, L.M.; Kehl, K.L.; Hassett, M.J.; Liu, D.; Schrag, D.; Gusev, A. Machine learning for genetics-based classification and treatment response prediction in cancer of unknown primary. Nat. Med. 2023, 29, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, Z.; Wang, W.; Huang, W.; Chen, C.; Xi, S.; Ahmad, M.U.; Ren, Y.; Sang, S.; Xie, J.; et al. Biology-guided deep learning predicts prognosis and cancer immunotherapy response. Nat. Commun. 2023, 14, 5135. [Google Scholar] [CrossRef]
- Mu, W.; Jiang, L.; Shi, Y.; Tunali, I.; Gray, J.E.; Katsoulakis, E.; Tian, J.; Gillies, R.J.; Schabath, M.B. Non-invasive measurement of PD-L1 status and prediction of immunotherapy response using deep learning of PET/CT images. J. Immunother. Cancer 2021, 9, e002118. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Yang, H.; Yeh, C.C.M.; Wang, L. Visual Analytics for RNN-Based Deep Reinforcement Learning. IEEE Trans. Vis. Comput. Graph. 2022, 28, 4141–4155. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hosny, A.; Zeleznik, R.; Parmar, C.; Coroller, T.; Franco, I.; Mak, R.H.; Aerts, H.J.W.L. Deep Learning Predicts Lung Cancer Treatment Response from Serial Medical Imaging. Clin. Cancer Res. 2019, 25, 3266–3275. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Islam, M.T.; Sang, S.; Qiu, L.; Xing, L. Biology-aware mutation-based deep learning for outcome prediction of cancer immunotherapy with immune checkpoint inhibitors. npj Precis. Oncol. 2023, 7, 117. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study | Name | Published Year | Countries | Cancer Type | Number of Participants | Drugs Used | Treatment Duration | Survival | Key Findings |
|---|---|---|---|---|---|---|---|---|---|
| Snyder et al. (2015) [69] | CheckMate 057: Nivolumab in Non-Small-Cell Lung Cancer (NSCLC) | 2015 | Global | NSCLC | 582 | Nivolumab, Docetaxel | Median follow-up of 18 months | 1-year: Nivolumab 51%, Docetaxel 39%; 18-month: Nivolumab 39%, Docetaxel 23% | Nivolumab significantly improved overall survival compared to Docetaxel in advanced non-squamous NSCLC after platinum-based chemotherapy. Nivolumab showed superior efficacy across all PD-L1 expression levels (≥1%, ≥5%, ≥10%). Lower incidence of grade 3–4 treatment-related adverse events in Nivolumab group (10% vs. 54%). |
| Reck et al. (2016) [74] | KEYNOTE-024: Pembrolizumab for NSCLC (First-Line Therapy) | 2016 | Global | NSCLC | 305 | Pembrolizumab, platinum-based chemotherapy | Median follow-up not specified, progression-free survival assessed | 6-month overall survival: Pembrolizumab 80.2%, Chemotherapy 72.4% | Pembrolizumab significantly improved progression-free and overall survival compared to chemotherapy in previously untreated advanced NSCLC with PD-L1 ≥50% expression and no EGFR or ALK mutations. Fewer grade 3–5 treatment-related adverse events in the Pembrolizumab group (26.6% vs. 53.3%). |
| Ferris et al. (2016) [75] | CheckMate 141: Nivolumab in Head and Neck Squamous Cell Carcinoma (HNSCC) | 2016 | Global (North America, Europe, Asia) | HNSCC | 361 | Nivolumab (anti-PD-1 monoclonal antibody) | Median follow-up not specified, progression-free survival assessed | Median overall survival: Nivolumab 7.5 months, standard therapy 5.1 months; 1-year survival: Nivolumab 36.0%, standard therapy 16.6% | Nivolumab significantly prolonged overall survival compared to standard therapy (hazard ratio 0.70, p = 0.01). The 1-year survival rate for Nivolumab was 19 percentage points higher. Median progression-free survival was 2.0 months for Nivolumab and 2.3 months for standard therapy. Nivolumab had a lower incidence of grade 3–4 treatment-related adverse events (13.1% vs. 35.1%) and maintained stable physical, role, and social functioning, while these worsened with standard therapy. |
| Emens et al. (2018) [76] | IMpassion130: Atezolizumab in Triple-Negative Breast Cancer (TNBC) | 2018 | International multicenter study | TNBC | 902 | Atezolizumab (anti-PD-L1 monoclonal antibody), Nab-paclitaxel | Median follow-up of 12.9 months | Median overall survival: Atezolizumab + Nab-paclitaxel 21.3 months, Placebo + Nab-paclitaxel 17.6 months | In the intention-to-treat population, median progression-free survival was 7.2 months for Atezolizumab + Nab-paclitaxel, compared to 5.5 months for placebo + Nab-paclitaxel. In the PD-L1-positive subgroup, progression-free survival was 7.5 months vs. 5.0 months. No new adverse effects were identified; adverse events leading to discontinuation occurred in 15.9% of the Atezolizumab group and 8.2% of the placebo group. |
| Rittmeyer et al. (2017) [77] | OAK Trial: Atezolizumab in NSCLC (Second-Line Therapy) | 2017 | 31 countries | NSCLC | 1225 | Atezolizumab, Docetaxel | Median follow-up of 12.6 months | Median overall survival (ITT): Atezolizumab 13.8 months, Docetaxel 9.6 months | Atezolizumab significantly improved overall survival compared to Docetaxel in previously treated NSCLC patients, with a favorable safety profile. |
| Robert et al. (2015) [78] | CheckMate 066: Nivolumab in Advanced Melanoma | 2015 | Not specified | Melanoma | 418 | Nivolumab, Dacarbazineprovided | Specific follow-up time not provided | 1-year overall survival: Nivolumab 72.9%, Dacarbazine 42.1% | Nivolumab significantly improved overall survival and progression-free survival compared to Dacarbazine in previously untreated patients with advanced melanoma without BRAF mutation. |
| Kaufman et al. (2016) [10] | Avelumab in Patients with Chemotherapy-Refractory Metastatic Merkel Cell Carcinoma | 2016 | North America, Europe, Australia, and Asia | MCC | 88 | Avelumab (Bavencio) | Median follow-up of 10.4 months | Objective response rate: 31.8% (28 of 88 patients, 95% CI 21.9–43.1) | Avelumab was associated with durable responses, most of which are still ongoing, and was well tolerated. Avelumab represents a new therapeutic option for advanced Merkel cell carcinoma. |
| Herbst et al. (2015) [79] | KEYNOTE-010: Pembrolizumab in Advanced NSCLC | 2015 | 24 countries | NSCLC | 1034 | Pembrolizumab, Docetaxel | Median overall survival: Pembrolizumab 2 mg/kg: 10.4 months, Pembrolizumab 10 mg/kg: 12.7 months, Docetaxel: 8.5 months | Overall survival significantly longer for Pembrolizumab 2 mg/kg (HR 0.71, 95% CI 0.58–0.88, p = 0.0008) and Pembrolizumab 10 mg/kg (HR 0.61, 95% CI 0.49–0.75, p < 0.0001) compared to Docetaxel | In PD-L1 ≥50% population, overall survival was significantly longer with Pembrolizumab (2 mg/kg: 14.9 months vs. Docetaxel 8.2 months, HR 0.54, p = 0.0002; 10 mg/kg: 17.3 months vs. Docetaxel 8.2 months, HR 0.50, p < 0.0001). Progression-free survival was also significantly longer with Pembrolizumab (2 mg/kg: 5.0 months vs. Docetaxel 4.1 months, HR 0.59, p = 0.0001; 10 mg/kg: 5.2 months vs. Docetaxel 4.1 months, HR 0.59, p < 0.0001). Grade 3–5 adverse events were less common with Pembrolizumab (13% for 2 mg/kg, 16% for 10 mg/kg) compared to Docetaxel (35%). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, W.; Sun, Q.; Xu, B.; Wang, R. Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review. Life 2025, 15, 283. https://doi.org/10.3390/life15020283
Tao W, Sun Q, Xu B, Wang R. Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review. Life. 2025; 15(2):283. https://doi.org/10.3390/life15020283
Chicago/Turabian StyleTao, Weichu, Qian Sun, Bingxiang Xu, and Ru Wang. 2025. "Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review" Life 15, no. 2: 283. https://doi.org/10.3390/life15020283
APA StyleTao, W., Sun, Q., Xu, B., & Wang, R. (2025). Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review. Life, 15(2), 283. https://doi.org/10.3390/life15020283