
ANXA2 Protein and Its Role in Neurodegeneration Processes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Structure of the ANXA2 Protein
3. Functions of the ANXA2 Protein
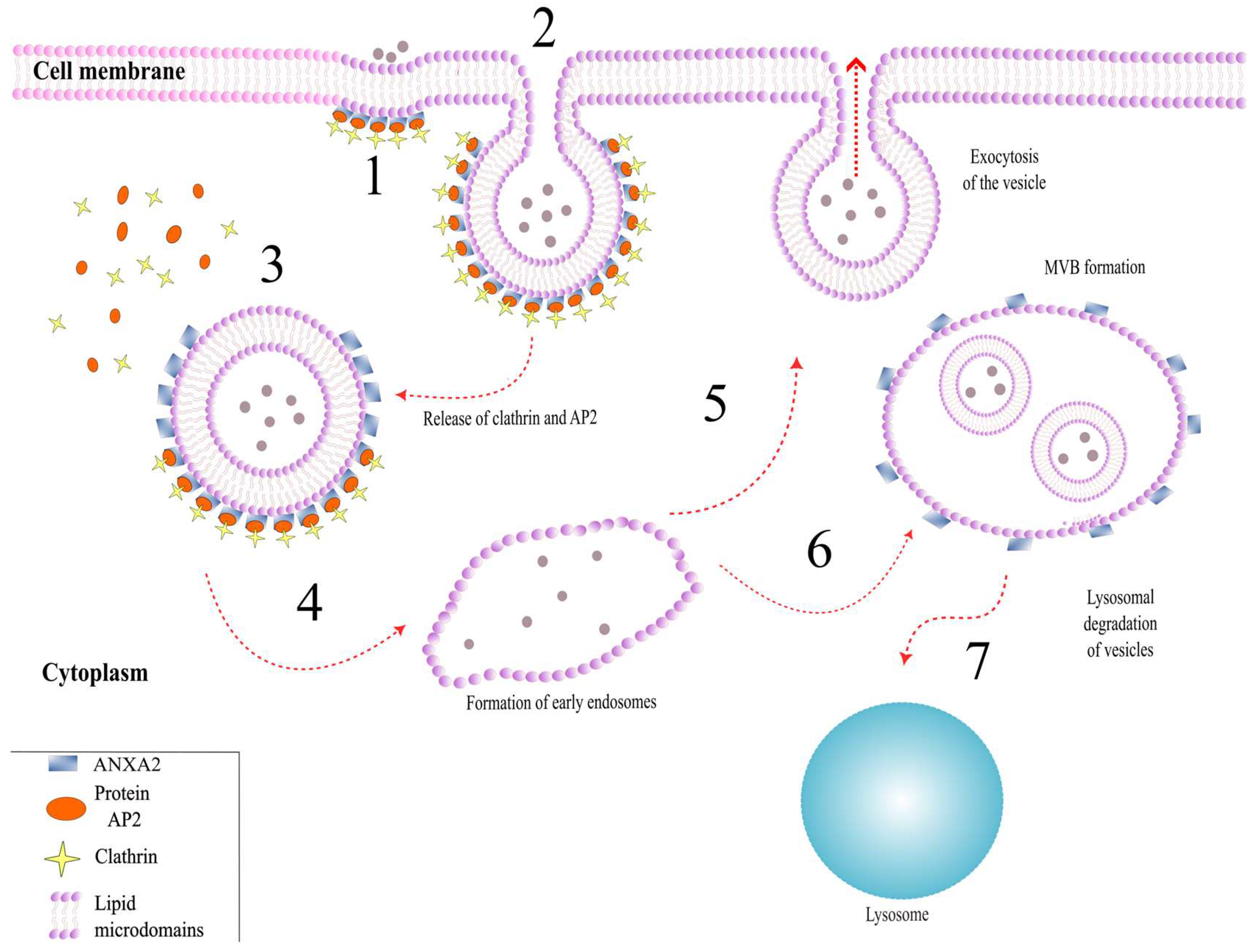
3.1. Role of ANXA2 in Exocytosis
3.2. Role of ANXA2 in Endocytosis
3.3. Role of ANXA2 in Antioxidant Protection of Cell
4. Involvement of ANXA2 in the Development of Neurodegenerative Processes
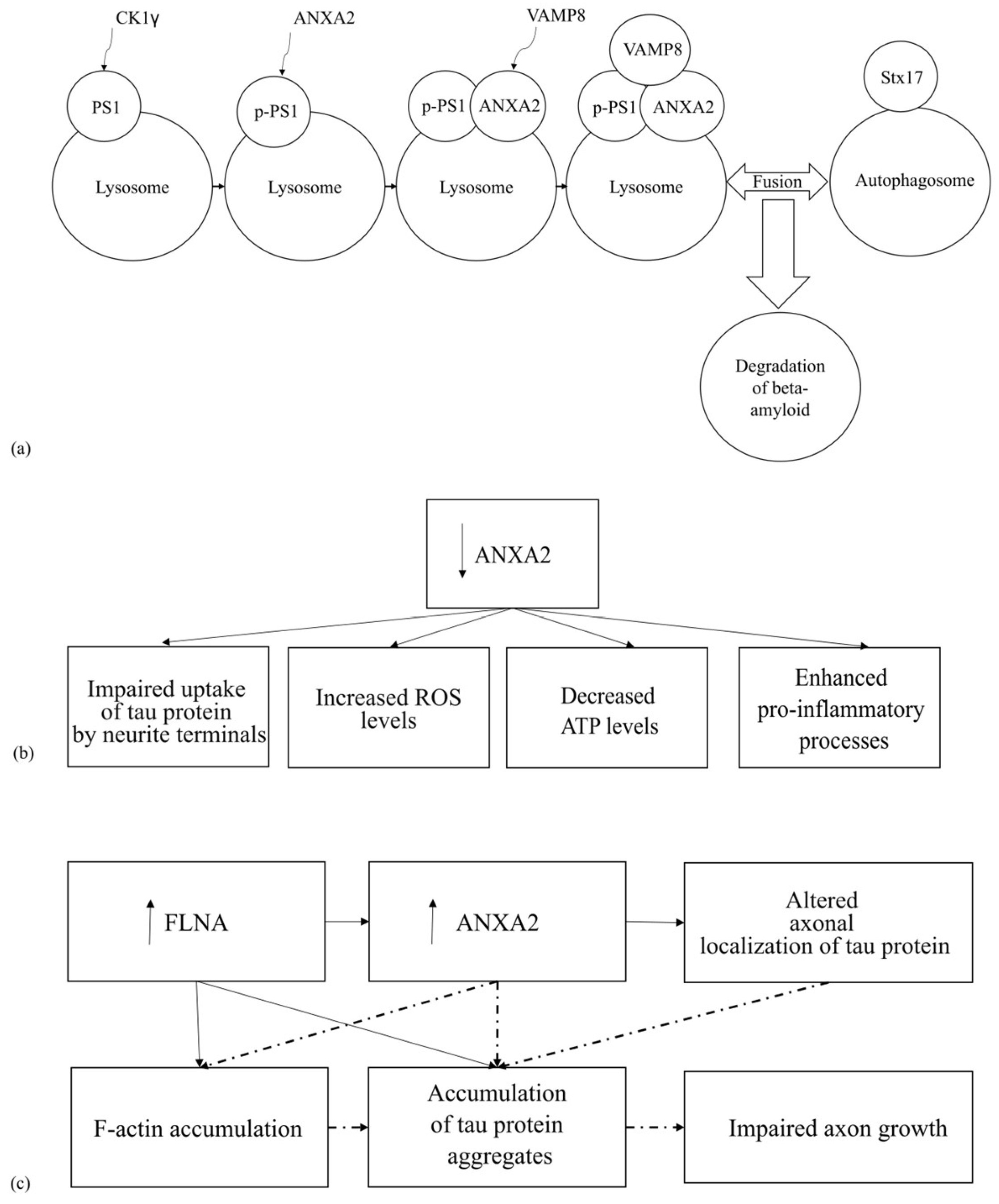
4.1. The Involvement of ANXA2 in the Pathogenesis of Tauopathies
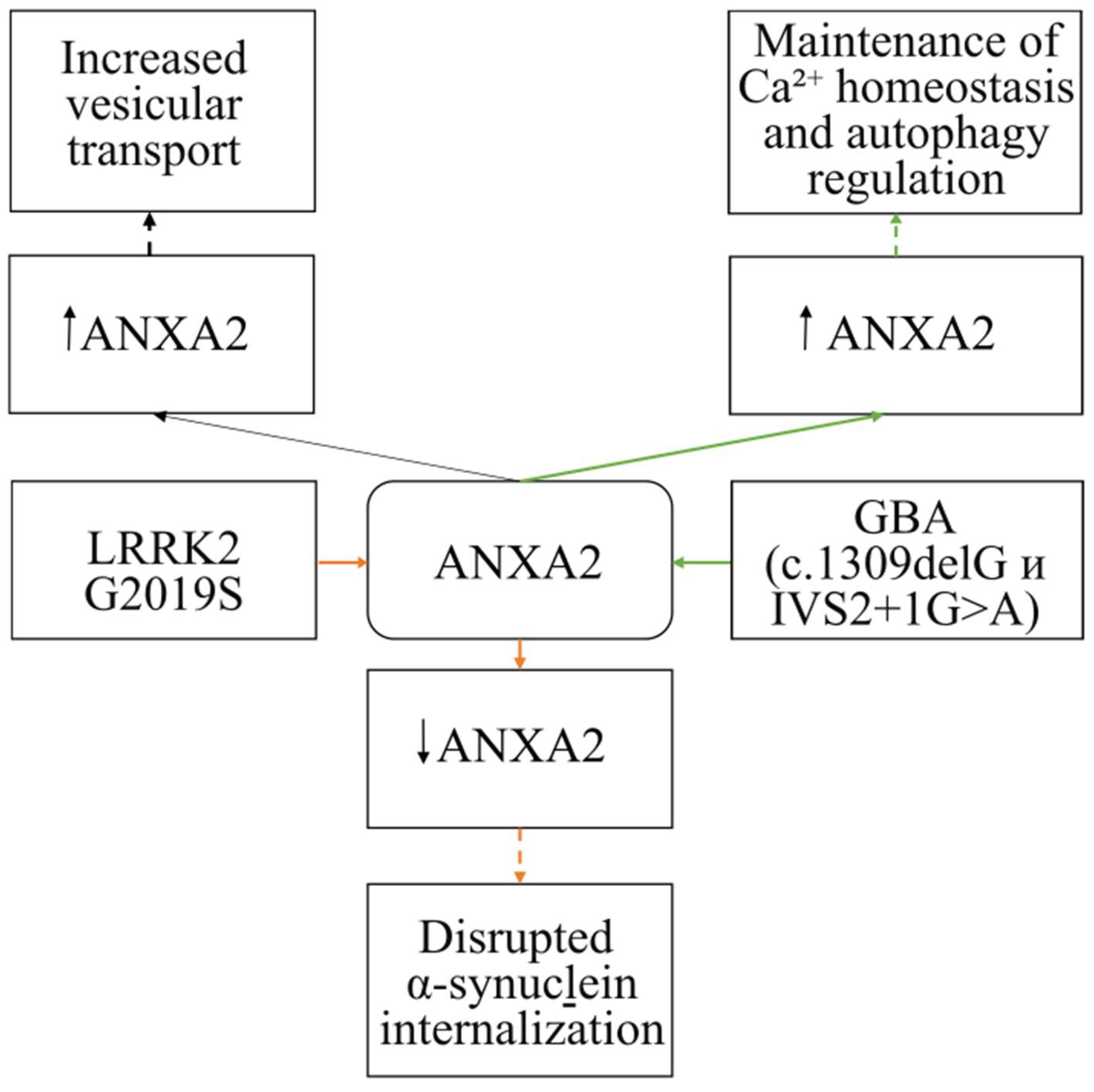
4.2. The Involvement of ANXA2 in the Pathogenesis of Parkinson’s Disease
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NDs | Neurodegenerative diseases |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| FTDP-17 | Frontotemporal dementia with parkinsonism |
| ANXA2 | Annexin A2 |
| tPA | tissue plasminogen activator |
| ROS | reactive oxygen species |
| APP | amyloid precursor protein |
References
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Ju, R.; Wang, Y. Roles of Annexin A protein family in autophagy regulation and therapy. Biomed. Pharmacother. 2020, 130, 110591. [Google Scholar] [CrossRef] [PubMed]
- White, Z.B., 2nd; Nair, S.; Bredel, M. The role of annexins in central nervous system development and disease. J. Mol. Med. 2024, 102, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, H.; Li, X.; Zhao, Y. Annexin A1 in the nervous and ocular systems. Neural Regen. Res. 2024, 19, 591–597. [Google Scholar] [CrossRef]
- Gauthier-Kemper, A.; Alonso, M.S.; Sündermann, F.; Niewidok, B.; Fernandez, M.-P.; Bakota, L.; Heinisch, J.J.; Brandt, R. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau’s axonal localization. J. Biol. Chem. 2018, 293, 8065–8076. [Google Scholar] [CrossRef]
- Streubel-Gallasch, L.; Giusti, V.; Sandre, M.; Tessari, I.; Plotegher, N.; Giusto, E.; Masato, A.; Iovino, L.; Battisti, I.; Arrigoni, G.; et al. Parkinson’s Disease-Associated LRRK2 Interferes with Astrocyte-Mediated Alpha-Synuclein Clearance. Mol. Neurobiol. 2021, 58, 3119–3140. [Google Scholar] [CrossRef]
- Jayaswamy, P.K.; Vijaykrishnaraj, M.; Patil, P.; Alexander, L.M.; Kellarai, A.; Shetty, P. Implicative role of epidermal growth factor receptor and its associated signaling partners in the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2023, 83, 101791. [Google Scholar] [CrossRef]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Castaldo, S.A.; Ajime, T.; Serrao, G.; Anastacio, F.; Rosa, J.T.; Giacomantonio, C.A.; Howarth, A.; Hill, R.; Madureira, P.A. Annexin A2 Regulates AKT Upon H2O2-Dependent Signaling Activation in Cancer Cells. Cancers 2019, 11, 492. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 heterotetramer: Structure and function. Int. J. Mol. Sci. 2013, 14, 6259–6305. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Kempster, E.; Waisman, D.M. The Annexin A2/S100A10 Complex: The Mutualistic Symbiosis of Two Distinct Proteins. Biomolecules 2021, 11, 1849. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Lin, C.F. Annexin A2: Its molecular regulation and cellular expression in cancer development. Dis. Markers 2014, 2014, 308976. [Google Scholar] [CrossRef] [PubMed]
- Grindheim, A.K.; Saraste, J.; Vedeler, A. Protein phosphorylation and its role in the regulation of Annexin A2 function. Biochim. Et Biophys. Acta (BBA)-General. Subj. 2017, 1861 Pt A, 2515–2529. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, J.C.; Martinez-Carmona, F.J.; Rodriguez-Crespo, I.; Lizarbe, M.A.; Turnay, J. Molecular dissection of the membrane aggregation mechanisms induced by monomeric annexin A2. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Raddum, A.M.; Hollas, H.; Shumilin, I.A.; Henklein, P.; Kretsinger, R.; Fossen, T.; Vedeler, A. The native structure of annexin A2 peptides in hydrophilic environment determines their anti-angiogenic effects. Biochem. Pharmacol. 2015, 95, 1–15. [Google Scholar] [CrossRef]
- Grindheim, A.K.; Patil, S.S.; Nebigil, C.G.; Desaubry, L.; Vedeler, A. The flavagline FL3 interferes with the association of Annexin A2 with the eIF4F initiation complex and transiently stimulates the translation of annexin A2 mRNA. Front. Cell Dev. Biol. 2023, 11, 1094941. [Google Scholar] [CrossRef]
- Jost, M.; Thiel, C.; Weber, K.; Gerke, V. Mapping of three unique Ca(2+)-binding sites in human annexin II. Eur. J. Biochem. 1992, 207, 923–930. [Google Scholar] [CrossRef]
- Johnsson, N.; Marriott, G.; Weber, K. p36, the major cytoplasmic substrate of src tyrosine protein kinase, binds to its p11 regulatory subunit via a short amino-terminal amphiphatic helix. EMBO J. 1988, 7, 2435–2442. [Google Scholar] [CrossRef]
- Morel, E.; Gruenberg, J. Annexin A2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J. Biol. Chem. 2009, 284, 1604–1611. [Google Scholar] [CrossRef]
- Gabel, M.; Delavoie, F.; Royer, C.; Tahouly, T.; Gasman, S.; Bader, M.-F.; Vitale, N.; Chasserot-Golaz, S. Phosphorylation cycling of Annexin A2 Tyr23 is critical for calcium-regulated exocytosis in neuroendocrine cells. Biochim. Et Biophys. Acta BBA-Mol. Cell Res. 2019, 1866, 1207–1217. [Google Scholar] [CrossRef]
- Umbrecht-Jenck, E.; Demais, V.; Calco, V.; Bailly, Y.; Bader, M.F.; Chasserot-Golaz, S. S100A10-mediated translocation of annexin-A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 2010, 11, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Gabel, M.; Delavoie, F.; Demais, V.; Royer, C.; Bailly, Y.; Vitale, N.; Bader, M.-F.; Chasserot-Golaz, S. Annexin A2–dependent actin bundling promotes secretory granule docking to the plasma membrane and exocytosis. J. Cell Biol. 2015, 210, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, K.T. Dominant role of lipid rafts L-type calcium channel in activity-dependent potentiation of large dense-core vesicle exocytosis. J. Neurochem. 2009, 110, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Bhatti, D.L.; Lee, K.-W.; Medrihan, L.; Cheng, J.; Wei, J.; Zhong, P.; Yan, Z.; Kooiker, C.; Song, C.; et al. Ahnak scaffolds p11/Anxa2 complex and L-type voltage-gated calcium channel and modulates depressive behavior. Mol. Psychiatry 2020, 25, 1035–1049. [Google Scholar] [CrossRef]
- Prada, I.; Cocucci, E.; Racchetti, G.; Meldolesi, J. The Ca2+-dependent exocytosis of enlargeosomes is greatly reinforced by genistein via a non-tyrosine kinase-dependent mechanism. FEBS Lett. 2007, 581, 4932–4936. [Google Scholar] [CrossRef]
- Lorusso, A.; Covino, C.; Priori, G.; Bachi, A.; Meldolesi, J.; Chieregatti, E. Annexin2 coating the surface of enlargeosomes is needed for their regulated exocytosis. EMBO J. 2006, 25, 5443–5456. [Google Scholar] [CrossRef]
- Benaud, C.; Gentil, B.J.; Assard, N.; Court, M.; Garin, J.; Delphin, C.; Baudier, J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 2004, 164, 133–144. [Google Scholar] [CrossRef]
- Borgonovo, B.; Cocucci, E.; Racchetti, G.; Podini, P.; Bachi, A.; Meldolesi, J. Regulated exocytosis: A novel, widely expressed system. Nat. Cell Biol. 2002, 4, 955–962. [Google Scholar] [CrossRef]
- Rezvanpour, A.; Santamaria-Kisiel, L.; Shaw, G.S. The S100A10-annexin A2 complex provides a novel asymmetric platform for membrane repair. J. Biol. Chem. 2011, 286, 40174–40183. [Google Scholar] [CrossRef]
- Cocucci, E.; Racchetti, G.; Rupnik, M.; Meldolesi, J. The regulated exocytosis of enlargeosomes is mediated by a SNARE machinery that includes VAMP4. J. Cell Sci. 2008, 121 Pt 18, 2983–2991. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Creutz, C.E.; Snyder, S.L. Interactions of annexins with the mu subunits of the clathrin assembly proteins. Biochemistry 2005, 44, 13795–13806. [Google Scholar] [CrossRef] [PubMed]
- Mettlen, M.; Chen, P.H.; Srinivasan, S.; Danuser, G.; Schmid, S.L. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 2018, 87, 871–896. [Google Scholar] [CrossRef] [PubMed]
- Drucker, P.; Pejic, M.; Galla, H.J.; Gerke, V. Lipid segregation and membrane budding induced by the peripheral membrane binding protein annexin A2. J. Biol. Chem. 2013, 288, 24764–24776. [Google Scholar] [CrossRef] [PubMed]
- Grieve, A.G.; Moss, S.E.; Hayes, M.J. Annexin A2 at the interface of actin and membrane dynamics: A focus on its roles in endocytosis and cell polarization. Int. J. Cell Biol. 2012, 2012, 852430. [Google Scholar] [CrossRef]
- Urbanska, A.; Sadowski, L.; Kalaidzidis, Y.; Miaczynska, M. Biochemical Characterization of APPL Endosomes: The Role of Annexin A2 in APPL Membrane Recruitment. Traffic 2011, 12, 1227–1241. [Google Scholar] [CrossRef]
- Mayran, N.; Parton, R.G.; Gruenberg, J. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. EMBO J. 2003, 22, 3242–3253. [Google Scholar] [CrossRef]
- Hayes, M.J.; Shao, D.M.; Grieve, A.; Levine, T.; Bailly, M.; Moss, S.E. Annexin A2 at the interface between F-actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochim. Biophys. Acta 2009, 1793, 1086–1095. [Google Scholar] [CrossRef]
- Morel, E.; Parton, R.G.; Gruenberg, J. Annexin A2-Dependent Polymerization of Actin Mediates Endosome Biogenesis. Dev. Cell 2009, 16, 445–457. [Google Scholar] [CrossRef]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef]
- Zobiack, N.; Rescher, U.; Ludwig, C.; Zeuschner, D.; Gerke, V. The annexin 2/S100A10 complex controls the distribution of transferrin receptor-containing recycling endosomes. Mol. Biol. Cell 2003, 14, 4896–4908. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Gruenberg, J. The p11/S100A10 light chain of annexin A2 is dispensable for annexin A2 association to endosomes and functions in endosomal transport. PLoS ONE 2007, 2, e1118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.R.; Fernandez, D.J.; Thornton, S.M.; Skeate, J.G.; Luhen, K.P.; Da Silva, D.M.; Langen, R.; Kast, W.M. Heterotetrameric annexin A2/S100A10 (A2t) is essential for oncogenic human papillomavirus trafficking and capsid disassembly, and protects virions from lysosomal degradation. Sci. Rep. 2018, 8, 11642. [Google Scholar] [CrossRef] [PubMed]
- Hedhli, N.; Falcone, D.J.; Huang, B.; Cesarman-Maus, G.; Kraemer, R.; Zhai, H.; Tsirka, S.E.; Santambrogio, L.; Hajjar, K.A. The Annexin A2/S100A10 System in Health and Disease: Emerging Paradigms. J. Biomed. Biotechnol. 2012, 2012, 406273. [Google Scholar] [CrossRef]
- Rentero, C.; Blanco-Munoz, P.; Meneses-Salas, E.; Grewal, T.; Enrich, C. Annexins-Coordinators of Cholesterol Homeostasis in Endocytic Pathways. Int. J. Mol. Sci. 2018, 19, 1444. [Google Scholar] [CrossRef]
- Sullivan, D.M.; Wehr, N.B.; Fergusson, M.M.; Levine, R.L.; Finkel, T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 2000, 39, 11121–11128. [Google Scholar] [CrossRef]
- Caplan, J.F.; Filipenko, N.R.; Fitzpatrick, S.L.; Waisman, D.M. Regulation of annexin A2 by reversible glutathionylation. J. Biol. Chem. 2004, 279, 7740–7750. [Google Scholar] [CrossRef]
- Madureira, P.A.; Hill, R.; Miller, V.A.; Giacomantonio, C.; Lee, P.W.; Waisman, D.M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget 2011, 2, 1075–1093. [Google Scholar] [CrossRef]
- Chen, L.; Lin, L.; Xian, N.; Zheng, Z. Annexin A2 regulates glioma cell proliferation through the STAT3-cyclin D1 pathway. Oncol. Rep. 2019, 42, 399–413. [Google Scholar] [CrossRef]
- Bharadwaj, A.G.; Kempster, E.; Waisman, D.M. The ANXA2/S100A10 Complex-Regulation of the Oncogenic Plasminogen Receptor. Biomolecules 2021, 11, 1772. [Google Scholar] [CrossRef]
- Sawangareetrakul, P.; Ngiwsara, L.; Champattanachai, V.; Chokchaichamnankit, D.; Saharat, K.; Ketudat Cairns, J.R.; Srisomsap, C.; Khwanraj, K.; Dharmasaroja, P.; Pulkes, T.J.B.R. Aberrant proteins expressed in skin fibroblasts of Parkinson’s disease patients carrying heterozygous variants of glucocerebrosidase and parkin genes. Biomed. Rep. 2021, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Creekmore, B.C.; Watanabe, R.; Lee, E.B. Neurodegenerative Disease Tauopathies. Annu. Rev. Pathol. 2023, 19, 345–370. [Google Scholar] [CrossRef] [PubMed]
- Donadio, V.; Sturchio, A.; Rizzo, G.; Rumeileh, S.A.; Liguori, R.; Espay, A.J. Pathology vs pathogenesis: Rationale and pitfalls in the clinicopathology model of neurodegeneration. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2023; Volume 192, pp. 35–55. [Google Scholar]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Weissmann, C.; Golovyashkina, N.; Sebo-Lemke, Z.; Drewes, G.; Gerke, V.; Heinisch, J.J.; Brandt, R. The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 2011, 192, 647–661. [Google Scholar] [CrossRef]
- Tsujikawa, K.; Hamanaka, K.; Riku, Y.; Hattori, Y.; Hara, N.; Iguchi, Y.; Ishigaki, S.; Hashizume, A.; Miyatake, S.; Mitsuhashi, S.; et al. Actin-binding protein filamin-A drives tau aggregation and contributes to progressive supranuclear palsy pathology. Sci. Adv. 2022, 8, eabm5029. [Google Scholar] [CrossRef]
- Burns, L.H.; Wang, H.Y. Altered filamin A enables amyloid beta-induced tau hyperphosphorylation and neuroinflammation in Alzheimer’s disease. Neuroimmunol. Neuroinflammation 2017, 4, 263–271. [Google Scholar] [CrossRef]
- Iwamoto, D.V.; Huehn, A.; Simon, B.; Huet-Calderwood, C.; Baldassarre, M.; Sindelar, C.V.; Calderwood, D.A. Structural basis of the filamin A actin-binding domain interaction with F-actin. Nat. Struct. Mol. Biol. 2018, 25, 918–927. [Google Scholar] [CrossRef]
- Levert, S.; Pilliod, J.; Aumont, E.; Armanville, S.; Tremblay, C.; Calon, F.; Leclerc, N. Direct and Indirect Effects of Filamin A on Tau Pathology in Neuronal Cells. Mol. Neurobiol. 2023, 60, 1021–1039. [Google Scholar] [CrossRef]
- Nizynski, B.; Dzwolak, W.; Nieznanski, K. Amyloidogenesis of Tau protein. Protein Sci. 2017, 26, 2126–2150. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Setu, J.R.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Role of PSEN Mutations in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2020, 38, 833–849. [Google Scholar] [CrossRef]
- Yepes, M. The plasminogen activating system in the pathogenesis of Alzheimer’s disease. Neural Regen. Res. 2021, 16, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Melchor, J.P.; Pawlak, R.; Strickland, S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J. Neurosci. 2003, 23, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bagyinszky, E.; An, S.S.A. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 8417. [Google Scholar] [CrossRef] [PubMed]
- Bustos, V.; Pulina, M.V.; Bispo, A.; Lam, A.; Flajolet, M.; Gorelick, F.S.; Greengard, P. Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome–lysosome fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 7148–7153. [Google Scholar] [CrossRef]
- Kang, H.M.; Choi, K.S.; Kassam, G.; Fitzpatrick, S.L.; Kwon, M.; Waisman, D.M. Role of annexin II tetramer in plasminogen activation. Trends Cardiovasc. Med. 1999, 9, 92–102. [Google Scholar] [CrossRef]
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325. [Google Scholar] [CrossRef]
- Ye, L.; Zhao, J.; Xiao, Z.; Gu, W.; Liu, X.; Ajuyo, N.M.C.; Min, Y.; Pei, Y.; Wang, D. Integrative Human Genetic and Cellular Analysis of the Pathophysiological Roles of AnxA2 in Alzheimer’s Disease. Antioxid. 2024, 13, 1274. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Rudenok, M.M.; Shadrina, M.I.; Filatova, E.V.; Rybolovlev, I.N.; Nesterov, M.S.; Abaimov, D.A.; Ageldinov, R.A.; Kolacheva, A.A.; Ugrumov, M.V.; Slominsky, P.A.; et al. Expression Analysis of Genes Involved in Transport Processes in Mice with MPTP-Induced Model of Parkinson’s Disease. Life 2022, 12, 751. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Partevian, S.A.; Slominsky, P.A.; Shadrina, M.I.; Alieva, A.K. ANXA2 Protein and Its Role in Neurodegeneration Processes. Life 2025, 15, 402. https://doi.org/10.3390/life15030402
Partevian SA, Slominsky PA, Shadrina MI, Alieva AK. ANXA2 Protein and Its Role in Neurodegeneration Processes. Life. 2025; 15(3):402. https://doi.org/10.3390/life15030402
Chicago/Turabian StylePartevian, Suzanna A., Petr A. Slominsky, Maria I. Shadrina, and Anelya Kh. Alieva. 2025. "ANXA2 Protein and Its Role in Neurodegeneration Processes" Life 15, no. 3: 402. https://doi.org/10.3390/life15030402
APA StylePartevian, S. A., Slominsky, P. A., Shadrina, M. I., & Alieva, A. K. (2025). ANXA2 Protein and Its Role in Neurodegeneration Processes. Life, 15(3), 402. https://doi.org/10.3390/life15030402