Is Struvite a Prebiotic Mineral?


Abstract
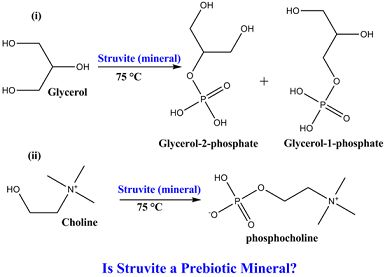
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Methods
2.3. Geochemical Modeling

3. Results
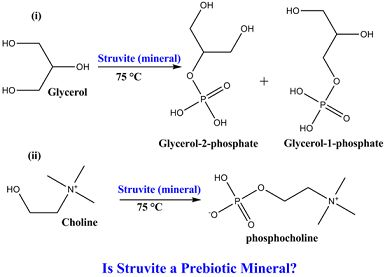
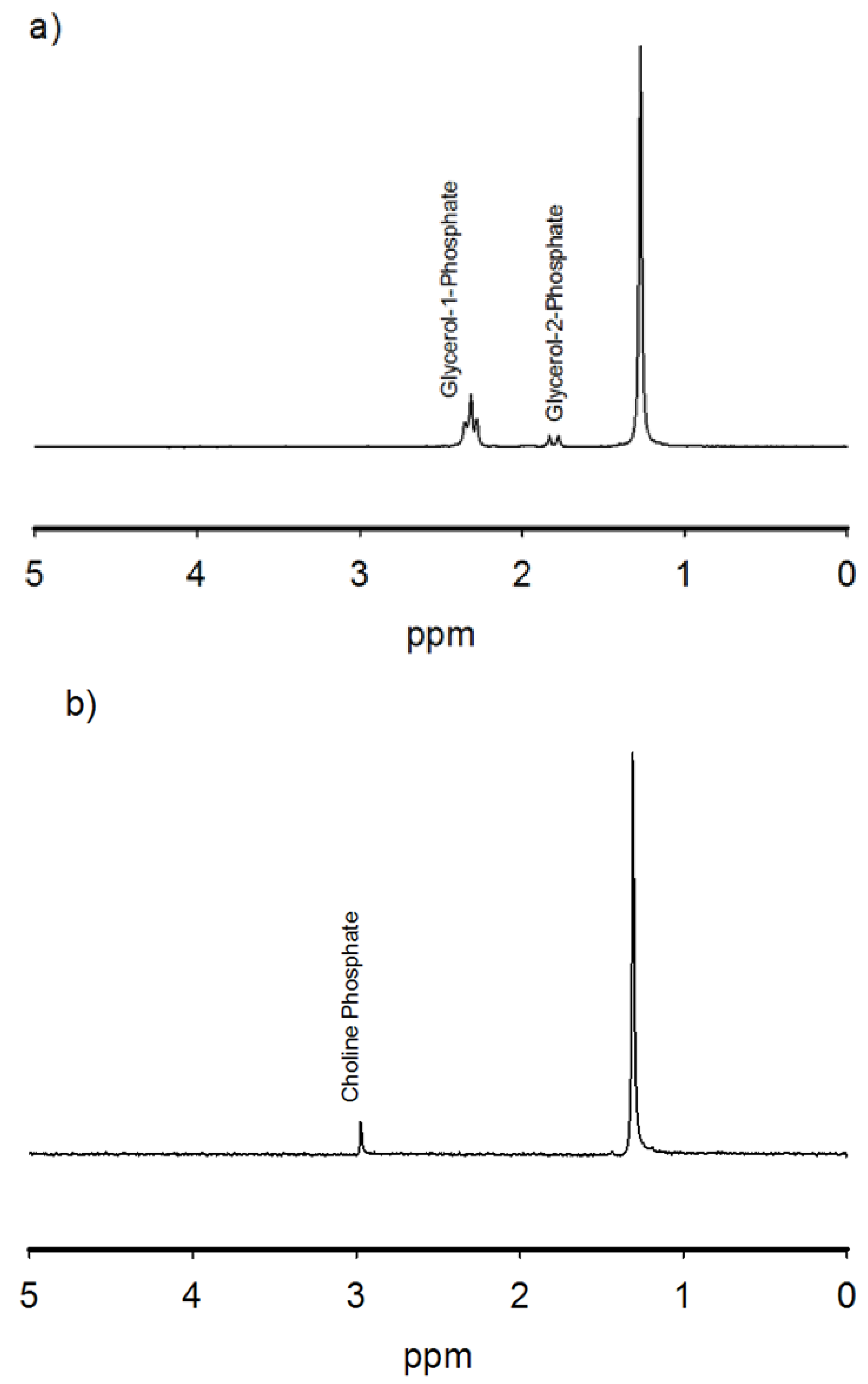
3.1. Experimental Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Yield (%) CH2-O-P | Yield (%)CH-OP |
|---|---|---|
| Glycerol + struvite | 28 ± 5 | 6 ± 2 |
| Choline chloride + struvite | 5 ± 1 | ------- |
| Glycerol + monetite | 12 ± 3 | BDL |
| Choline chloride + monetite | BDL | BDL |

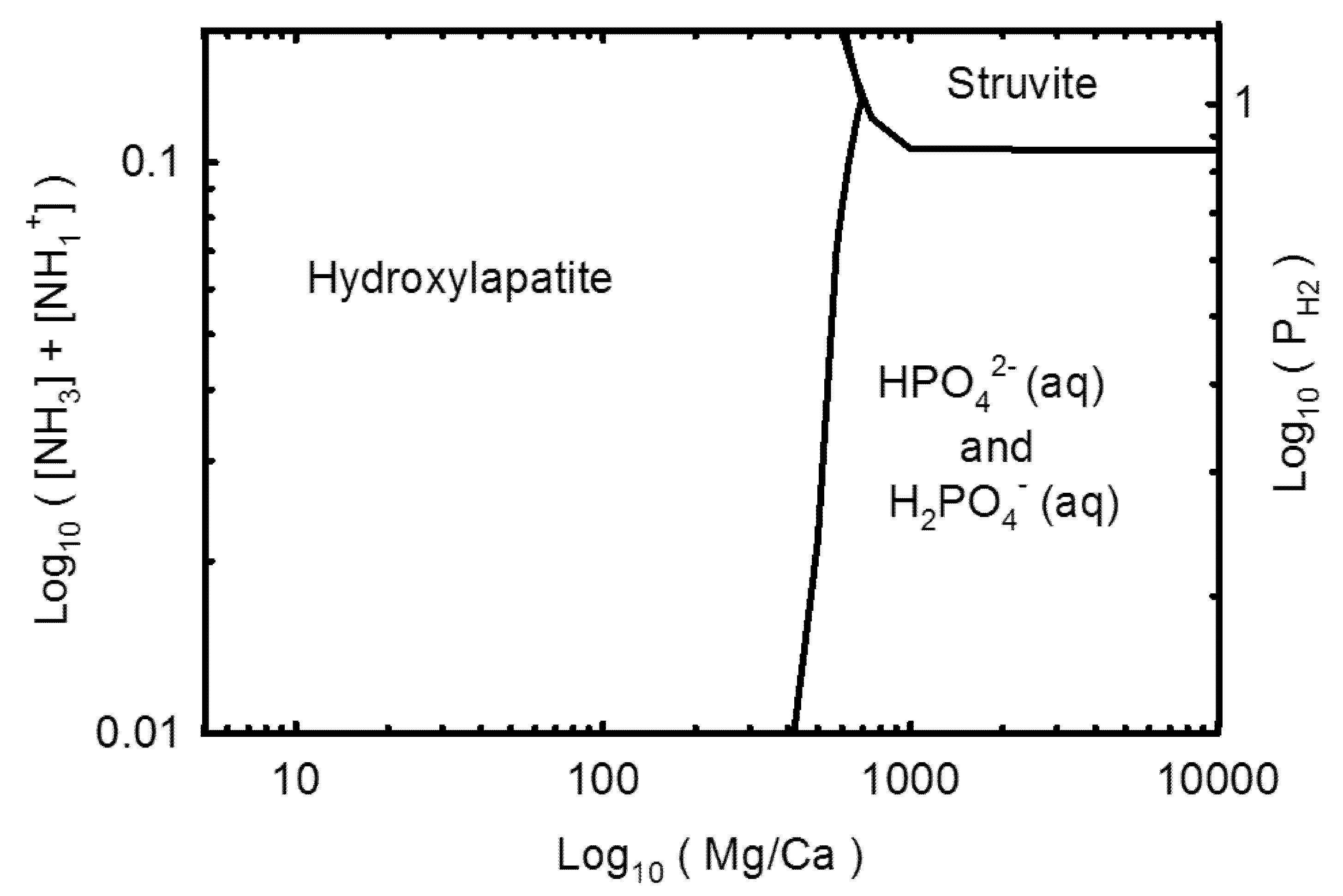
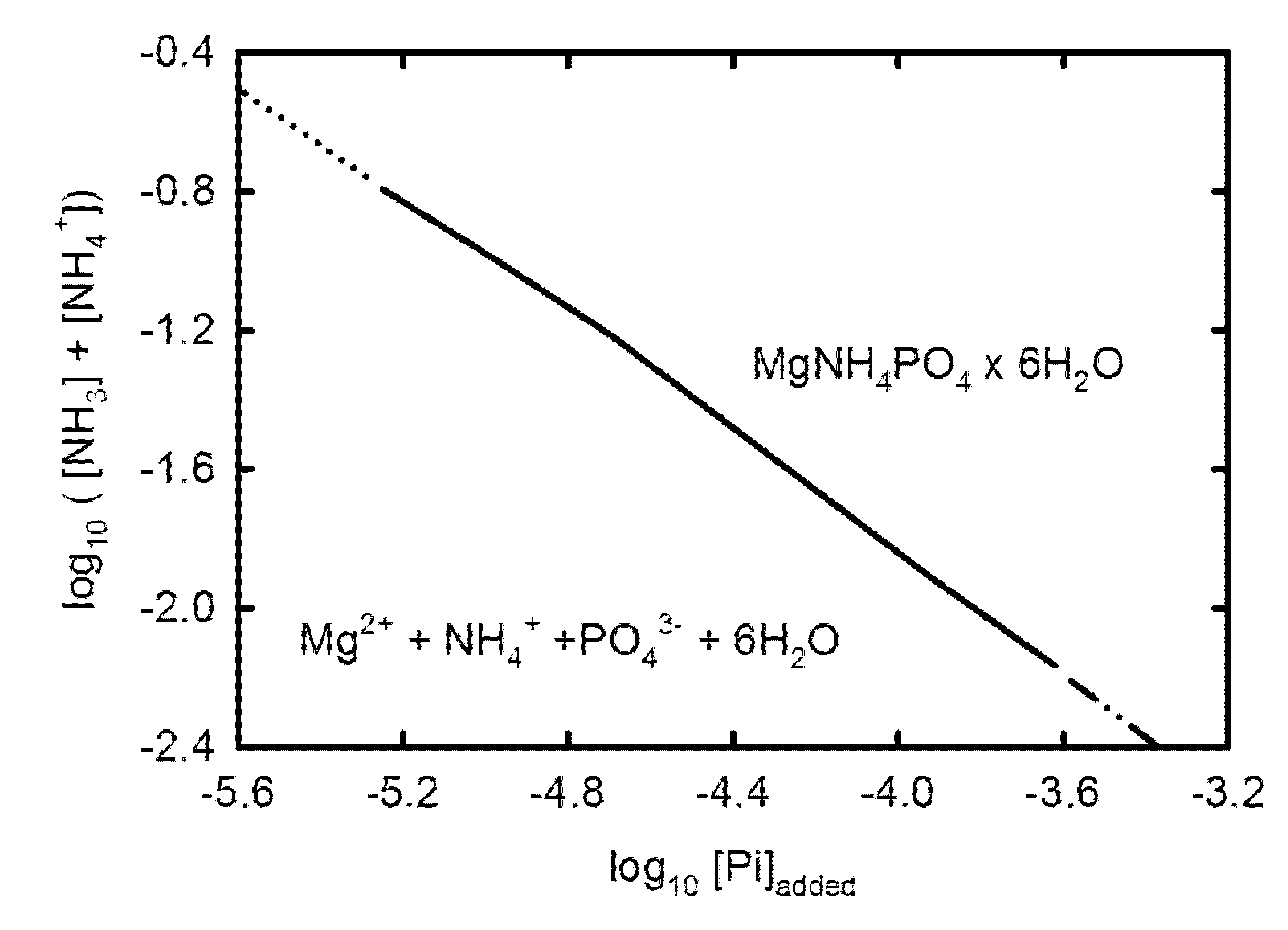
3.2. Geochemical Modeling


4. Discussion
5. Conclusions
Acknowledgments
References
- Pasek, M.A. Rethinking early Earth phosphorus geochemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 853–858. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P. On the origin of phosphorylated biomolecules. In Origins of Life: The Primal Self-Organization; Egel, R., Lankenau, D.-H., Mulkidjanian, A.Y., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany, 2011; pp. 57–84. [Google Scholar]
- Arrhenius, G.; Sales, B.; Mojzsis, S.; Lee, T. Entropy and charge in molecular evolution-the case of phosphate. J. Theor. Biol. 1997, 187, 503–522. [Google Scholar] [CrossRef]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Philos. Trans. Roy. Soc. B 2006, 361, 1743–1749. [Google Scholar] [CrossRef]
- Gulick, A. Phosphorus as a factor in the origin of life. Am. Sci. 1955, 43, 479–489. [Google Scholar]
- Gedulin, B.; Arrhenius, G. Sources and geochemical evolution of RNA precursor molecules—The role of phosphate. In Early Life on Earth, Nobel Symposium; Bengston, S., Ed.; Columbia University Press: New York, NY, USA, 1994; Volume 84, pp. 91–110. [Google Scholar]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar]
- Hazen, R.M.; Papineau, D.; Bleeker, W.; Downs, R.T.; Ferry, J.M.; McCoy, T.J.; Sverjensky, D.A.; Yang, H. Mineral evolution. Am. Mineral. 2008, 93, 1693–1720. [Google Scholar] [CrossRef]
- Handschuh, G.J.; Orgel, L.E. Struvite and prebiotic phosphorylation. Science 1973, 179, 483–484. [Google Scholar]
- Ferris, J.P.; Nicodem, D.E. Ammonia photolysis and the Role of ammonia in chemical revolution. Nature 1972, 238, 268–269. [Google Scholar] [CrossRef]
- Smirnov, A.; Hausner, D.; Laffers, R.; Strongin, D.R.; Schoonen, M.A.A. Abiotic ammonium formation in the presence of Ni-Fe metals and alloys and its implications for the Hadean nitrogen cycle. Geochem. Trans. 2008, 9, 1–20. [Google Scholar] [CrossRef]
- Hargreaves, W.R.; Mulvihill, S.J.; Deamer, D.W. Synthesis of phospholipids and membranes in prebiotic conditions. Nature 1977, 266, 78–80. [Google Scholar] [CrossRef]
- Gull, M.; Yu, W.; Yingwu, W.; Zhan, S.; Ge, T.; Shouhua, F. Mimicking the prebiotic acidic hydrothermal environment: One-pot prebiotic hydrothermal synthesis of glucose phosphates. Heteroat. Chem. 2011, 22, 186–191. [Google Scholar] [CrossRef]
- Gull, M.; Ge, T.; Yingwu, W.; Chao, H.; Zhan, S.; Hongming, Y.; Shouhua, F. Resolving the enigma of prebiotic C-O-P bond formation: Prebiotic hydrothermal synthesis of important biological phosphate esters. Heteroat. Chem. 2010, 21, 161–167. [Google Scholar]
- HSC Chemistry Program Web Site. Available online: https://www.hsc-chemistry.net/ (accessed on 15 April 2013).
- White, W.B.; Johnson, S.M.; Dantzig, G.B. Chemical equilibrium in complex mixtures. J. Chem. Phys. 1958, 28, 751–755. [Google Scholar] [CrossRef]
- Pasek, M.A.; Lauretta, D.S. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early earth. Astrobiology 2005, 5, 515–535. [Google Scholar] [CrossRef]
- Pasek, M.A.; Greenberg, R. Acidification of Europa’s subsurface ocean as a consequence of oxidant delivery. Astrobiology 2012, 12, 151–159. [Google Scholar] [CrossRef]
- Pasek, M.A.; Block, K.; Pasek, V. Fulgurite morphology: A classification scheme and clues to formation. Contrib. Mineral. Petrol. 2012, 164, 477–492. [Google Scholar] [CrossRef]
- Bhuiyan, M.I.; Mavinic, D.S.; Beckie, R.D. A solubility and thermodynamic study of struvite. Environ. Technol. 2007, 28, 1015–1026. [Google Scholar] [CrossRef]
- Kim, D.; Ryu, H.-D.; Kim, M.-S.; Kim, J.; Lee, S.-I. Enhancing struvite precipitation potential for ammonia nitrogen removal in municipal landfill leachate. J. Hazard. Mater. 2006, 146, 81–85. [Google Scholar]
- Bouropoulos, N.C.; Koutsoukos, P.G. Spontaneous precipitation of struvite from aqueous solutions. J. Cryst. Growth 2000, 213, 381–388. [Google Scholar] [CrossRef]
- Martens, C.S.; Harriss, R.C. Inhibition of apatite precipitation in the marine environment by magnesium ion. Geochim. Cosmochim. Acta 1970, 34, 621–625. [Google Scholar] [CrossRef]
- Tatur, A.; Keck, A. Phosphates in Ornithogenic soils of the maritime Antarctic. Proc. NIPR Symp. Polar Biol. 1990, 3, 133–150. [Google Scholar]
- Konhauser, K.O.; Lalonde, S.V.; Amskold, L.; Holland, H.D. Was there really an Archean phosphate crisis? Science 2007, 315, 1234. [Google Scholar] [CrossRef]
- Paytan, A.; McLaughlin, K. The oceanic phosphorus cycle. Chem. Rev. 2007, 107, 563–576. [Google Scholar] [CrossRef]
- Pasek, M.A.; Lauretta, D.S. Extraterrestrial flux of potentially prebiotic C, N, and P to the early Earth. Orig. Life Evol. Biospheres 2008, 38, 5–21. [Google Scholar] [CrossRef]
- Cullen, D.J. Mineralogy of nitrogenous guano on the Bounty islands, SW Pacific ocean. Sedimentology 1988, 35, 421–428. [Google Scholar] [CrossRef]
- Griffith, D.P. Struvite stones. Kidney Int. 1978, 13, 372–382. [Google Scholar] [CrossRef]
- Booker, N.A.; Priestley, A.J.; Fraser, I.H. Struvite formation in waste water treatment plants opportunities for nutrient recovery. Environ. Technol. 1999, 20, 777–782. [Google Scholar] [CrossRef]
- Pasek, M.A.; Dworkin, J.P.; Lauretta, D.S. A radical pathway for organic phosphorylation during Schreibersite corrosion with implications for the origin of life. Geochim. Cosmochim. Acta 2007, 71, 1721–1736. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gull, M.; Pasek, M.A. Is Struvite a Prebiotic Mineral? Life 2013, 3, 321-330. https://doi.org/10.3390/life3020321
Gull M, Pasek MA. Is Struvite a Prebiotic Mineral? Life. 2013; 3(2):321-330. https://doi.org/10.3390/life3020321
Chicago/Turabian StyleGull, Maheen, and Matthew A. Pasek. 2013. "Is Struvite a Prebiotic Mineral?" Life 3, no. 2: 321-330. https://doi.org/10.3390/life3020321
APA StyleGull, M., & Pasek, M. A. (2013). Is Struvite a Prebiotic Mineral? Life, 3(2), 321-330. https://doi.org/10.3390/life3020321