
The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells

Abstract
:1. Introduction
2. Autocatalysis in the Origin of Life
3. Energetics, Genetics, Catalysis and Membranes
4. Thioesters Were Used for Synthesis in Primitive Cells
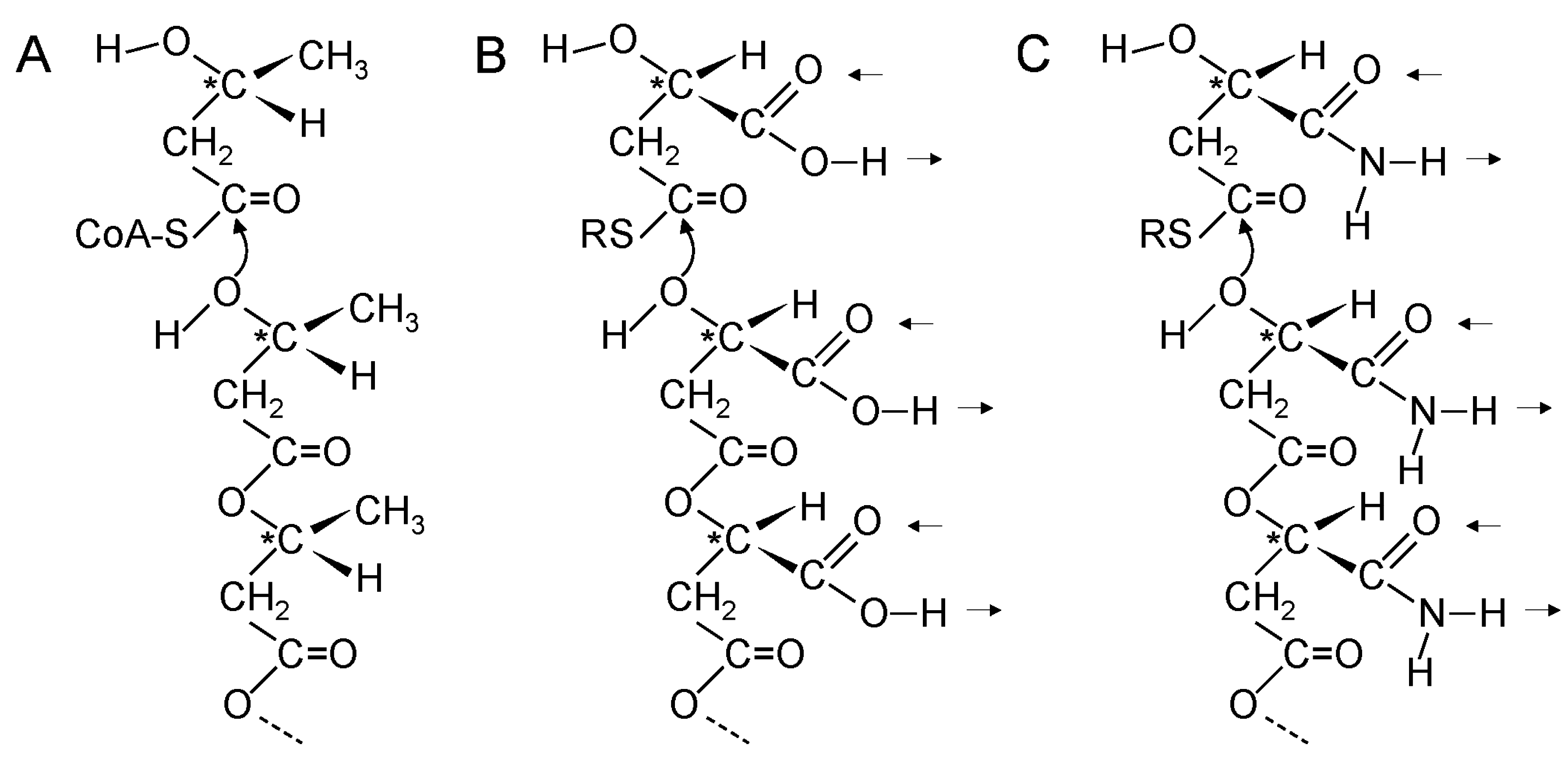
5. Thioester Bonds Were Used to Form Ester and Amide Bonds
6. Energy Transduction in Biology
7. Formation of the Earliest Cell Membranes
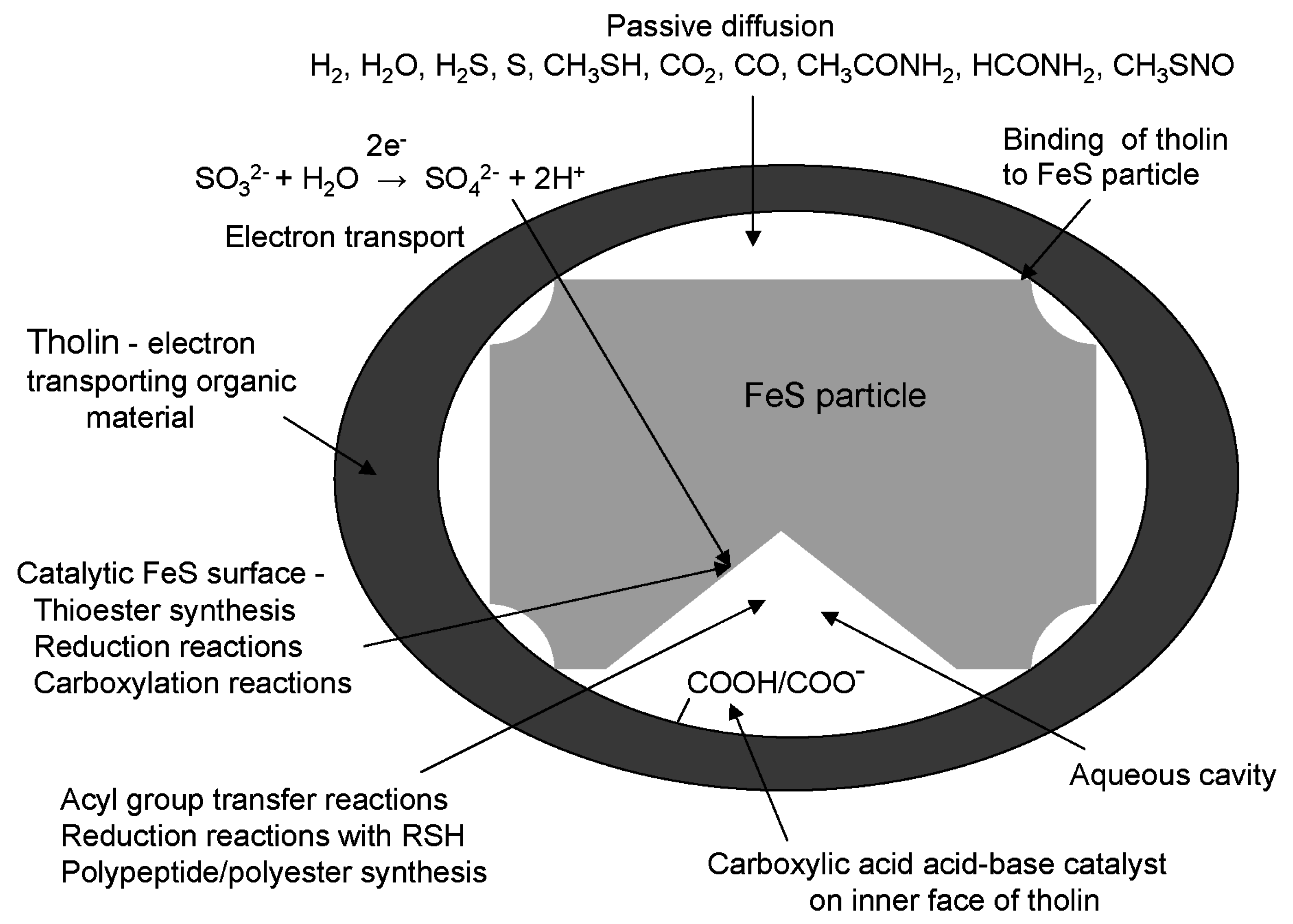
8. Energization of Primitive Cells Using Tholin as an Electron Transporting Organic Membrane
9. Iron Sulfide in Origin of Life Scenarios
10. FeS Cluster Biochemistry and Chemistry
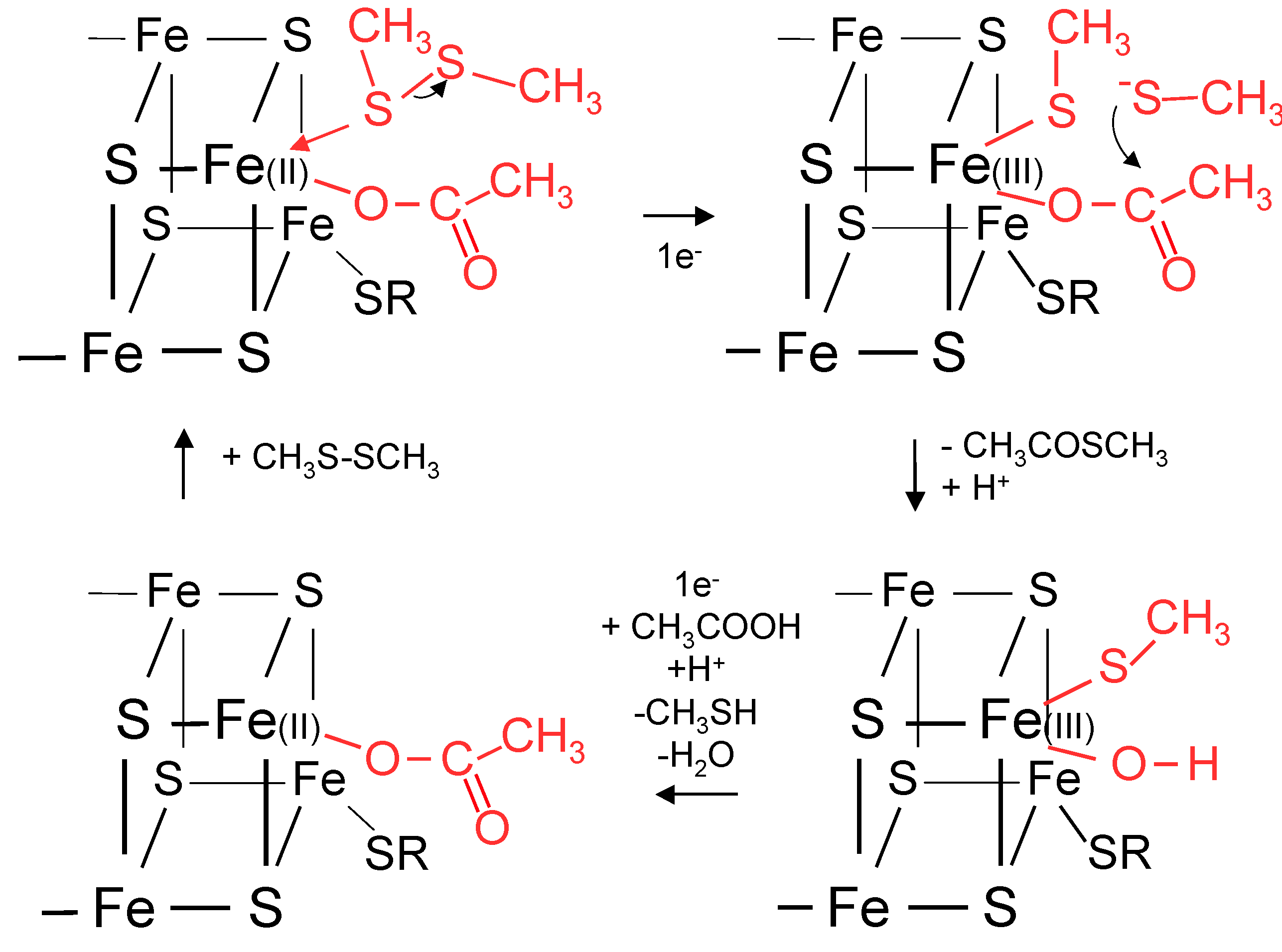
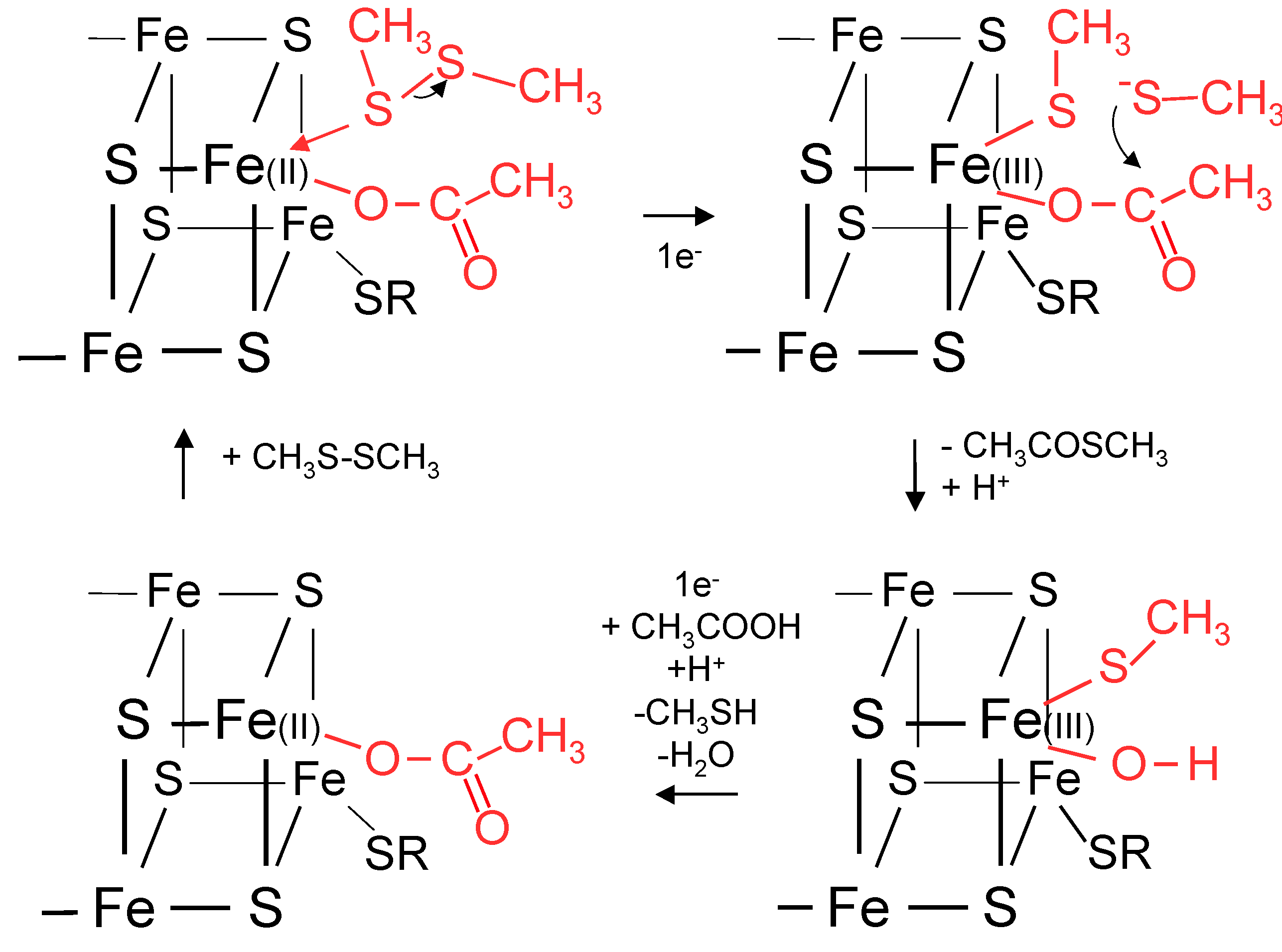
11. Biochemical Reduction of Disulfides and Thioester Formation
12. A Proposal for Primitive Cellular Synthesis of Thioesters
13. Diffusion of Small Polar and Nonpolar Molecules across Primitive Cell Membranes
14. Reduction and Carboxylation Reactions inside Primitive Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
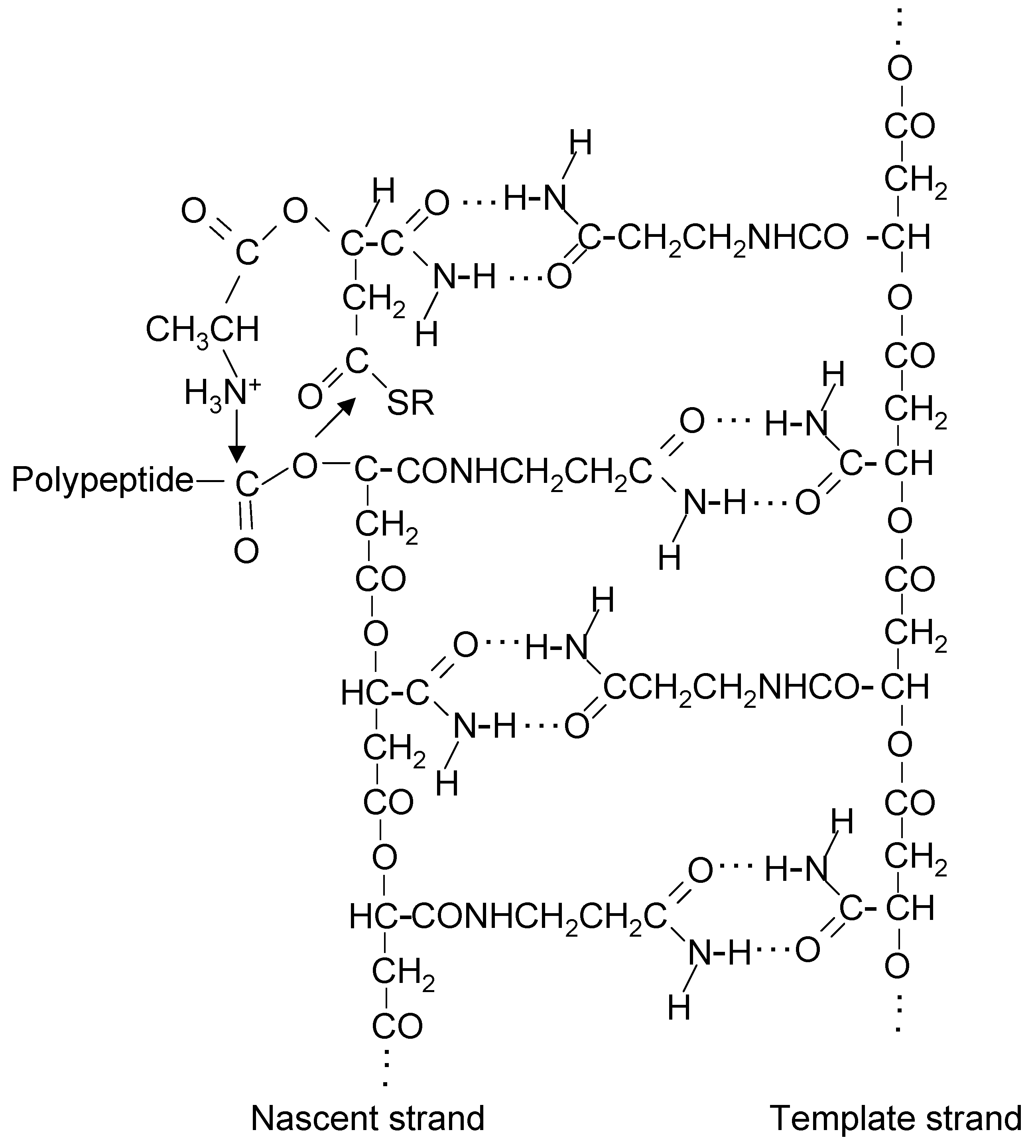{kind=link}
| Activity | Reaction |
|---|---|
| 1. Thioester synthesis | CH3COOH + 2e− + 2H+ + RSSR → CH3COSR + RSH + H2O |
| 2. CO reduction | CO + 2e− + 2H+ → H2CO |
| 3. CO2 reduction | CO2 + 2H+ + 2e− → HCOOH |
| 4. Nitrite reduction | NO2− + 8H+ + 6e− → NH4+ + 2H2O |
| 5. Reductive carboxylation | CH3COSR + 2e− + CO2 + 2H+ → CH3COCOOH + RSH |
| 6. Reductive amination | CH3COCOOH + 2RSH + NH3 → CH3CH(NH2)COOH + RSSR + H2O |
| CH3COCOOH + 2e− + 2H+ + NH3 → CH3CH(NH2)COOH + H2O | |
| 7. Glycine synthesis | H2CO + NH3 + CO2 + 2RSH → HOOCCH2NH2 + H2O + RSSR |
| 8. Claisen condensation | CH3COSR + HCOCOOH → HOOCCH(OH)CH2COSR |
| 9. Carboxylation | CH3COCOOH + CO2 → HOOCCH2COCOOH |
| 10. Reduction of ketone | HOOCCOCH2COOH + 2e− + 2H+ → HOOCCH(OH)CH2COOH |
| 11. Reductive thiolation | HCOCOOH + 2e− + H2S + 2H+ → HSCH2COOH + H2O |
| 12. Aldol condensation | 2CH3COCOOH → HOOCC(CH3)OHCH2COCOOH |
| 13. Acyl group transfer | R’COSR + R”OH → R’COOR” + HSR |
| R’COSR + R”NH2 → R’CONHR” + HSR | |
| 14. β-Polyester synthesis | –(COCH2CH(COOH)O)n– + RSCOCH2CH(COOH)OH → –(COCH2CH(COOH)O)n+1– + RSH |
| 15. Polypeptide synthesis | –(NHCHR’CO)n– + H2NCHR’COSR → –(NHCHR’CO)n+1– + HSR |
15. Base Catalysis in Early Metabolism
16. A Model for the Simple System within a Primitive Cell

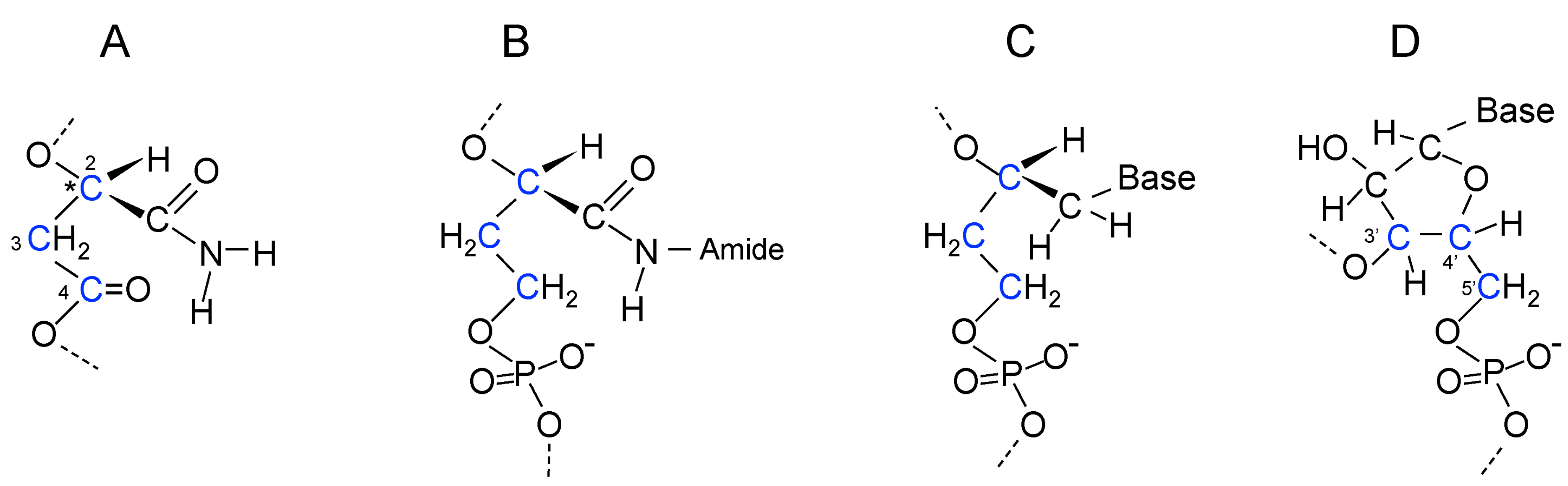
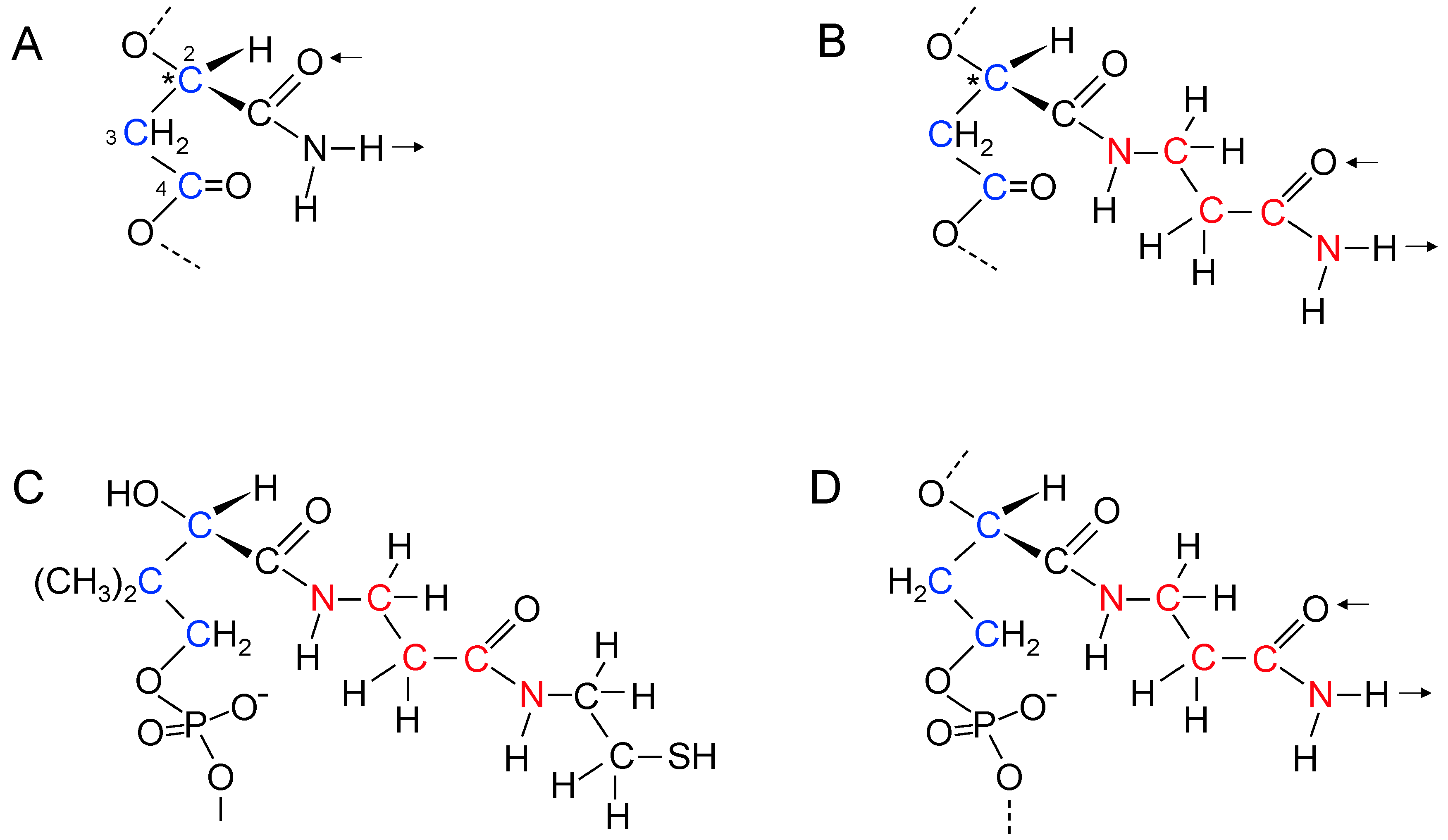
17. Genetic Polymers Were Initially Produced from Aliphatic Monomers


18. Evolution of the Purines and Pyrimidines of Nucleic Acids from Aliphatic Side Chains


19. Coenzyme A as a Relic of Early Nucleic Acid Evolution
20. Coupling of β-Polyester Synthesis and Protein Synthesis

21. A Thiol at the N-Terminal End of Early Proteins
22. Autocatalysis Revisited
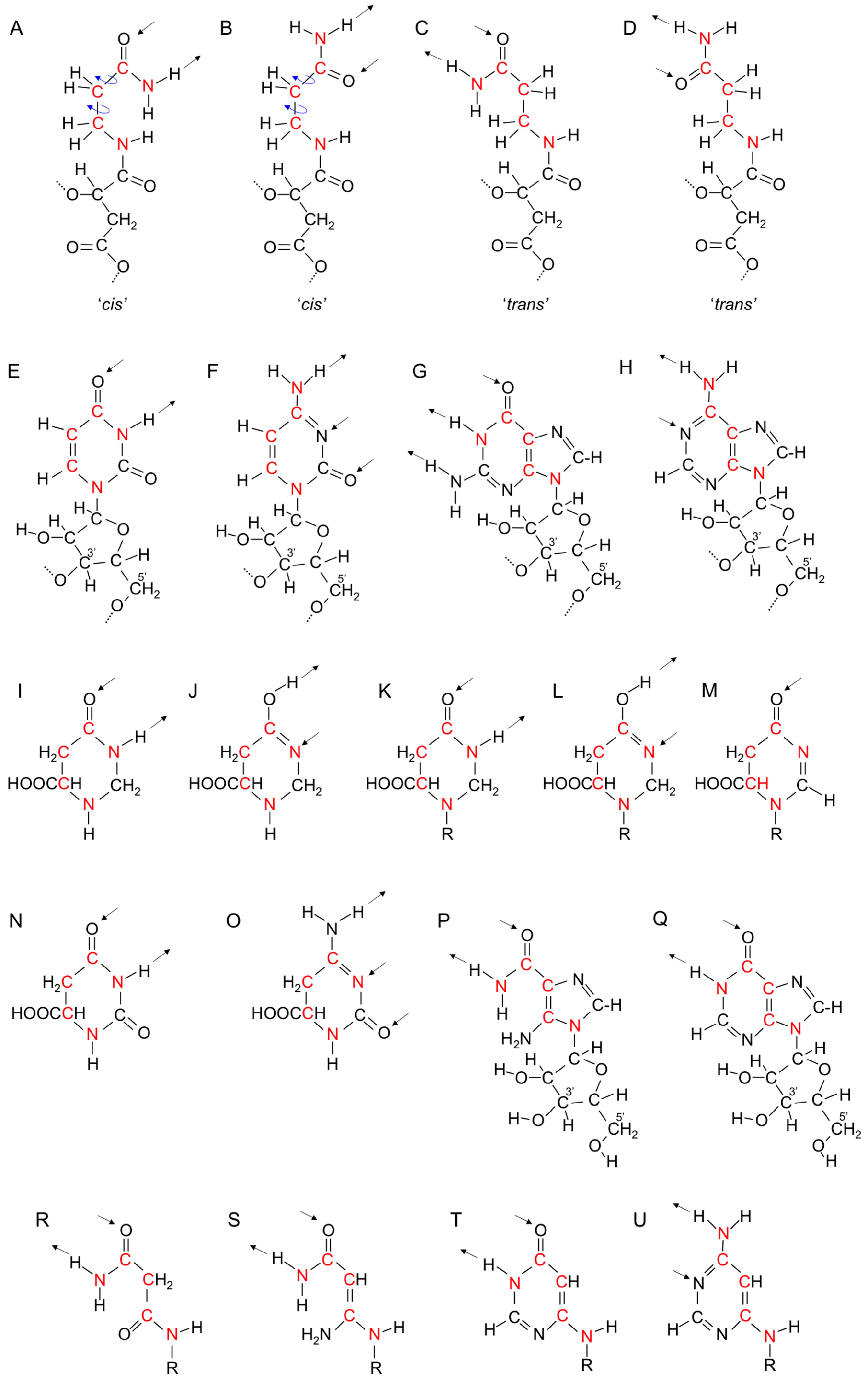
23. Replication of β-Polyesters
24. Evolution of the Singlet Coding System into the Triplet Coding System
25. Overview of the Evolution of Nucleic Acid Synthesis, and the Origin and Evolution of the Genetic Code
- (1)
- (2)
- (3)
- (4)
- (5)
- (6)
- (7)
- (8)
- Codons: GUC codes for Val, GCC codes for Ala, GAC codes for Asp, GGC codes for Gly. The second base in the codon, underlined, is the same as the base in the singlet coding system.
- (9)
- Expansion of genetic code to include 20 amino acids.
26. Discussion
26.1. An Evolutionary Pathway from Primitive Cells to Biology
26.2. Comparisons with Other Proposals on the Origins of Cells
26.3. Why Is the Simple Method of Thioester Synthesis Not Used in Biology?
26.4. Coded Proteins Enabled Evolution of Nucleic Acid and Ribosomal Protein Synthesis
26.5. The Need for Simplicity of Metabolism, Catalysis, and Replication in the Primitive Cell
26.6. Reversible Reactions in Primitive Cells
26.7. The Four Bases in Nucleic Acids Are Derived from the Four Orientations of Carboxamide Side Chains
26.8. Did “Chemical Evolution” Happen before Biological Evolution?
Conflicts of Interest
References
- Neveu, M.; Kim, H.-J.; Benner, S.A. The “strong” RNA world hypothesis: Fifty years old. Astrobiology 2013, 13, 391–403. [Google Scholar]
- Joyce, G.F. The antiquity of RNA-based evolution. Nature 2002, 418, 214–221. [Google Scholar]
- Kurland, C.G. The RNA dreamtime. Bioessays 2010, 32, 866–871. [Google Scholar]
- Bernhardt, H.S. The RNA world hypothesis: The worst theory of the early evolution of life (except for all the others). Biol. Direct 2012, 7. [Google Scholar] [CrossRef]
- Francis, B.R. An alternative to the RNA world hypothesis. Trends Evol. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Li, L.; Francklyn, C.; Carter, C.W. Aminoacylating urzymes challenge the RNA world hypothesis. J. Biol. Chem. 2013, 288, 26856–26863. [Google Scholar]
- Caetano-Anollés, G.; Seufferheld, M.J. The coevolutionary roots of biochemistry and cellular organization challenge the RNA world paradigm. J. Mol. Microbiol. Biotechnol. 2013, 23, 152–177. [Google Scholar]
- Francis, B.R. A hypothesis that ribosomal protein synthesis evolved from coupled protein and nucleic acid synthesis. Chemtracts Biochem. Mol. Biol. 2000, 13, 153–191. [Google Scholar]
- Lipmann, F. Attempts to map a process evolution of peptide biosynthesis. Science 1971, 173, 875–885. [Google Scholar]
- Wächtershäuser, G. Evolution of the first metabolic cycles. Proc. Natl. Acad. Sci. USA 1990, 87, 200–204. [Google Scholar]
- Morowitz, H.J.; Kostelnik, J.D.; Yang, J.; Cody, G.D. The origin of intermediary metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 7704–7798. [Google Scholar]
- Smith, E.; Morowitz, H.J. Universality in intermediary metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 13168–13173. [Google Scholar]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–363. [Google Scholar]
- Hazen, R.; Sverjensky, D.A. Mineral surfaces, geochemical complexities, and the origins of life. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Trefil, J.; Morowitz, H.; Smith, E. The Origin of Life. Am. Sci. 2009, 97. [Google Scholar] [CrossRef]
- Nitschke, W.; Russell, M.J. Beating the acetyl coenzyme A-pathway to the origin of life. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef]
- Sousa, F.L.; Thiergart, T.; Landan, G.; Nelson-Sathi, S.; Pereira, I.A.; Allen, J.F.; Lane, N.; Martin, W.F. Early bioenergetic evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef]
- Weber, A.L. Nonenzymatic formation of “energy rich” lactoyl and glyceroyl thioesters from glyceraldehyde and a thiol. J. Mol. Evol. 1984, 20, 157–166. [Google Scholar]
- Weber, A.L. Prebiotic amino acid thioester synthesis: Thiol-dependent amino acid synthesis from formose substrates (formaldehyde and glycoaldehyde) and ammonia. Orig. Life Evol. Biosph. 1998, 28, 259–270. [Google Scholar]
- De Duve, C. Blueprint for a Cell: The Nature and Origin of Life; Neil Patterson Publishers: Burlington, NC, USA, 1991. [Google Scholar]
- Wächtershäuser, G. Groundworks for an evolutionary biology: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar]
- Heinen, W.; Lauwers, A.-M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic environment. Orig. Life Evol. Biosph. 1996, 26, 131–150. [Google Scholar]
- Baltscheffsky, H.; Schultz, A.; Baltscheffsky, M. Energy for the origin of life. In Exobiology: Matter, Energy, and Information in the Origin and Evolution of Life in the Universe; Chela-Flores, J., Raulin, F., Eds.; Kluwer Academic Publishers: South Holland, The Netherlands, 1998; pp. 95–102. [Google Scholar]
- Deamer, D.W.; Weber, A.L. Bioenergetics and life’s origins. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Koonin, E.V. The origins of cellular life. Antonie Van Leeuwenhoek 2014, 106, 27–41. [Google Scholar]
- Janssen, M.J. Thiolo, thiono, and dithio acids and esters. In The Chemistry of Carboxylic Acids and Esters; Patai, S., Ed.; Interscience: London, UK, 1969; pp. 705–764. [Google Scholar]
- Wieland, T.; Schäfer, W. Synthese von oligopeptiden unter zellmoglichen bedingungen. Angew. Chem. 1951, 63, 146–147. [Google Scholar]
- Wieland, T.; Pfleiderer, G. Activation of amino acids. Adv. Enzymol. Relat. Subj. Biochem. 1957, 19, 235–266. [Google Scholar]
- Maurel, M.C.; Orgel, L.E. Oligomerization of α-thioglutamic acid. Orig. Life Evol. Biosph. 2000, 30, 423–430. [Google Scholar]
- Zepik, H.H.; Rajamani, S.; Maurel, M.C.; Deamer, D. Oligomerization of thioglutamic acid: Encapsulated reactions and lipid catalysis. Orig. Life Evol. Biosph. 2007, 37, 495–505. [Google Scholar]
- Weber, A.L. Oligoglyceric acid synthesis by autocondensation of glyceroyl thioester. J. Mol. Evol. 1987, 25, 191–196. [Google Scholar]
- Dawes, E.A.; Senior, P.J. The role and regulation of energy reserve polymers in micro-organisms. Adv. Microbiol. Physiol. 1973, 10, 135–266. [Google Scholar]
- Castresana, J.; Saraste, M. Evolution of energetic metabolism: The respiration-early hypothesis. Trends Biochem. Sci. 1995, 20, 443–448. [Google Scholar]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar]
- Liu, Y.; Beer, L.L.; Whitman, W.B. Sulfur metabolism in archaea reveals novel processes. Environ. Microbiol. 2012, 14, 2632–2644. [Google Scholar]
- Khare, B.N.; Thompson, W.R.; Chyba, C.F.; Arakawa, E.T.; Sagan, C. Organic solids produced from simple C/H/O/N ices by charged particles: Applications to the outer solar system. Adv. Space Res. 1989, 9, 41–53. [Google Scholar]
- Cody, G.D.; Boctor, N.Z.; Filley, T.R.; Hazen, R.M.; Scott, J.H.; Yoder, H.S., Jr. Primordial carbonylated iron-sulfur compounds and the synthesis of pyruvate. Science 2000, 289, 1337–1340. [Google Scholar]
- Yin, S.; Wang, Z.; Bernstein, E.R. Formaldehyde and methanol formation from reaction of carbon monoxide and hydrogen on neutral Fe2S2 clusters in the gas phase. Phys. Chem. Chem. Phys. 2013, 15, 4699–4706. [Google Scholar]
- Fuchs, G. Alternative pathways of carbon dioxide fixation: Insights into the early evolution of life? Ann. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar]
- O’Connell, C.; Hommeltoft, S.I.; Eisenberg, R. Electrochemical approaches to the reduction of carbon dioxide. In Carbon Dioxide as a Source of Carbon; Aresta, M., Forti, G., Eds.; D. Reidel Publishing Co.: Dordrecht, The Netherlands, 1987; pp. 33–54. [Google Scholar]
- Tezuka, M.; Yajima, T.; Tsuchiya, A.; Matsumoto, Y.; Uchida, Y.; Hidai, M. Electroreduction of carbon dioxide catalyzed by iron-sulfur cluster compounds [Fe4S4(SR)4]2−. J. Am. Chem. Soc. 1982, 104, 6834–6836. [Google Scholar]
- Lucas, B.H.; Ritcey, G.M. Reduction of ferric iron by SO2 with heat or SO2 with activated carbon. In Department of Energy, Mines and Resources, Technical Bulletin TB 107; Queen’s Printer and Controller of Stationary: Ottawa, ON, Canada, 1969. [Google Scholar]
- Cremlyn, R.J. An Introduction to Organosulfur Chemistry; John Wiley & Sons: Chichester, UK, 1996. [Google Scholar]
- Ducluzeau, A.L.; van Lis, R.; Duval, S.; Schoepp-Cothenet, B.; Russell, M.J.; Nitschke, W. Was nitric oxide the first deep electron sink? Trends Biochem. Sci. 2009, 34, 9–15. [Google Scholar]
- Mancinelli, R.L.; McKay, C.P. The evolution of nitrogen cycling. Orig. Life Evol. Biosph. 1988, 18, 311–325. [Google Scholar]
- Summers, D.P.; Chang, S. Prebiotic ammonia from reduction of nitrite by iron(II) on the early Earth. Nature 1993, 365, 630–633. [Google Scholar]
- Summers, D.P. Ammonia formation by the reduction of nitrite/nitrate by FeS: Ammonia formation under acidic conditions. Orig. Life Evol. Biosph. 2005, 35, 299–312. [Google Scholar]
- Summers, D.P.; Basa, R.C.; Khare, B.; Rodoni, D. Abiotic nitrogen fixation on terrestrial planets: Reduction of NO to ammonia by FeS. Astrobiology 2012, 12, 107–114. [Google Scholar]
- Saville, B. A scheme for colorimetric determination of microgram amounts of thiol. Analyst 1958, 83, 670–672. [Google Scholar]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-nitrosoglutathione. Biochim. Biophys. Acta. 2013, 1830, 3173–3181. [Google Scholar]
- Chyba, C.E.; Sagan, C. Endogenous production, exogenous delivery, and impact-shock synthesis of organic molecules: An inventory for the origin of life. Nature 1992, 355, 125–132. [Google Scholar]
- Deamer, D.W. Origins of life: How leaky were primitive cells. Nature 2008, 454, 37–38. [Google Scholar]
- Mansy, S.S. Model protocells from single-chain lipids. Int. J. Mol. Sci. 2009, 10, 835–843. [Google Scholar]
- Chen, I.A.; Walde, P. From self-assembled vesicles to protocells. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Pizzarello, S.; Shock, E. The organic composition of carbonaceous meteorites: The evolutionary story ahead of biochemistry. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Allamandola, L.J.; Sandford, S.A.; Wpoenka, B. Interstellar polycyclic aromatic hydrocarbons and carbon in interplanetary dust particles and meteorites. Science 1987, 237, 56–59. [Google Scholar]
- Parker, E.T.; Zhou, M.; Burton, A.S.; Glavin, D.P.; Dworkin, J.P.; Krishnamurthy, R.; Fernandez, F.M.; Bada, J.L. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew. Chem. Int. Ed. Engl. 2014, 53, 8132–8136. [Google Scholar]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of first cells at terrestrial, anoxic geothermal fields. Proc. Natl. Acad. Sci. USA 2012, 109, E821–E830. [Google Scholar]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Open questions on the origin of life at anoxic geothermal fields. Orig. Life Evol. Biosph. 2012, 42, 507–516. [Google Scholar]
- Parker, E.T.; Cleaves, H.J.; Dworkin, J.P.; Glavin, D.P.; Callahan, M.; Aubrey, A.; Lazcano, A.; Bada, J.L. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl. Acad. Sci. USA 2011, 108, 5526–5531. [Google Scholar]
- Sagan, C.; Khare, B.N. Tholins: Organic chemistry of interstellar grains and gas. Nature 1979, 277, 102–107. [Google Scholar]
- McCord, T.B.; Carlson, R.W.; Smythe, W.D.; Hansen, G.B.; Clark, R.N.; Hibbitts, C.A.; Fanale, F.P.; Granahan, J.C.; Segura, M.; Matson, D.L.; et al. Organics and other molecules in the surfaces of Callisto and Ganymede. Science 1997, 278, 271–275. [Google Scholar]
- Khare, B.N.; Bakes, E.L.; Cruikshank, D.; McKay, C.P. Solid organic matter in the atmosphere and on the surface of outer Solar System bodies. Adv. Space Res. 2001, 27, 299–307. [Google Scholar]
- Waite, J.H., Jr.; Young, D.T.; Cravens, T.E.; Coates, A.J.; Crary, F.J.; Magee, B.; Westlake, J. The process of tholin formation in Titan’s upper atmosphere. Science 2007, 316, 870–875. [Google Scholar]
- Gu, X.; Kim, Y.S.; Kaiser, R.I.; Mebel, A.M.; Liang, M.C.; Yung, Y.L. Chemical dynamics of triacetylene formation and implications to the synthesis of polyynes in Titan’s atmosphere. Proc. Natl. Acad. Sci. USA 2009, 106, 16078–16083. [Google Scholar]
- Folsom, C.E. The Origin of Life; W.H. Freeman & Co.: San Francisco, CA, USA, 1979. [Google Scholar]
- McDonald, G.D.; Khare, B.N.; Thompson, W.R.; Sagan, C. CH4/NH3/H2O spark tholin: Chemical analysis and interaction with Jovian aqueous clouds. Icarus 1991, 94, 354–367. [Google Scholar]
- Khare, B.N.; Sagan, C. Synthesis of cystine in simulated primitive conditions. Nature 1971, 232, 577–579. [Google Scholar]
- Van Trump, J.E.; Miller, S.L. Prebiotic synthesis of methionine. Science 1972, 178, 859–860. [Google Scholar]
- Raulin, F.; Toupance, G. Effect of H2S on the formation of organic compounds from C, N, H, S model atmospheres submitted to a glow discharge. Orig. Life 1975, 6, 507–512. [Google Scholar]
- Khare, B.N.; Sagan, C.; Bandurski, E.L.; Nagy, B. Ultraviolet-photoproduced organic solids synthesized under simulated jovian conditions: Molecular analysis. Science 1978, 199, 1199–1201. [Google Scholar]
- Minard, R.D.; Hatcher, P.G.; Gourley, R.C.; Matthews, C.N. Structural investigations of hydrogen cyanide polymers: New insights using TMAH thermochemolysis/GC-MS. Orig. Life Evol. Biosph. 1998, 28, 461–473. [Google Scholar]
- He, C.; Lin, G.; Upton, K.T.; Imanaka, H.; Smith, M.A. Structural investigation of Titan tholins by solution-state 1H, 13C, and 15N NMR: One-dimensional and decoupling experiments. J. Phys. Chem. A 2012, 116, 4760–4767. [Google Scholar]
- Martin, W.F.; Sousa, F.L.; Lane, N. Evolution. Energy at life’s origin. Science 2014, 344, 1092–1093. [Google Scholar]
- Bernstein, M.P.; Sandford, S.A.; Allamandola, L.J.; Gillette, J.S.; Clemett, S.J.; Zare, R.N. UV irradiation of polycyclic aromatic hydrocarbons in ices: Production of alcohols, quinones, and ethers. Science 1999, 283, 1135–1138. [Google Scholar]
- Hayashi, T.; Stuchebrukhov, A.A. Electron tunneling in respiratory complex I. Proc. Natl. Acad. Sci. USA 2010, 107, 19157–19162. [Google Scholar]
- Schenning, A.P.; Meijer, F.W. Supramolecular electronics; nanowires from self-assembled π-conjugated systems. Chem. Commun. (Camb.) 2005. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Electron transfer in proteins. Annu. Rev. Biochem. 1996, 65, 537–561. [Google Scholar]
- Seyedsayamdost, M.R.; Yee, C.S.; Reece, S.Y.; Nocera, D.G.; Stubbe, J. pH Rate profiles of FnY356-R2s (n = 2, 3, 4) in Escherichia coli ribonucleotide reductase: Evidence that Y356 is a redox-active amino acid along the radical propagation pathway. J. Am. Chem. Soc. 2006, 128, 1562–1568. [Google Scholar]
- Koch, A.L.; Schmidt, T.M. The first cellular bioenergetic process: Primitive generation of a proton-motive force. J. Mol. Evol. 1991, 33, 297–304. [Google Scholar]
- Wächtershäuser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar]
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997, 276, 245–247. [Google Scholar]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar]
- Eck, R.V.; Dayhoff, M.O. Evolution of the structure of ferredoxin based on living relics of primitive amino acid sequences. Science 1966, 152, 363–366. [Google Scholar]
- Davis, B.K. Evolution before the origin of species. Prog. Biophys. Mol. Biol. 2002, 79, 77–133. [Google Scholar]
- Evans, M.C.; Buchanan, B.B. Photoreduction of ferredoxin and its use in carbon dioxide fixation by a subcellular system from a photosynthetic bacterium. Proc. Natl. Acad. Sci. USA 1965, 53, 1420–1425. [Google Scholar]
- Reed, G.H.; Ragsdale, S.W.; Mansoorabadi, S.O. Radical reactions of thiamin pyrophosphate in 2-oxoacid oxidoreductases. Biochim. Biophys. Acta 2012, 1824, 1291–1298. [Google Scholar]
- Flint, D.H.; Allen, R.M. Iron-sulfur proteins with nonredox functions. Chem. Rev. 1996, 96, 2315–2334. [Google Scholar]
- Van Vugt-Lussenburg, B.M.; van der Weel, L.; Hagen, W.R.; Hagedoorn, P.L. Biochemical similarities and differences between the catalytic [4Fe-4S] cluster containing fumarases FumA and FumB from Escherichia coli. PLoS One 2013, 8, e55549. [Google Scholar]
- Holm, R.H. Synthetic approaches to the active sites of iron-sulfur proteins. Acc. Chem. Res. 1977, 10, 427–434. [Google Scholar]
- Holm, R.H. Electron transfer: Iron-sulfur clusters. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: New York, NY, USA, 2003; Volume 8, pp. 61–90. [Google Scholar]
- Bonomi, F.; Werth, M.T.; Kurtz, D.M. Assembly of FenSn(SR)2− (n = 2, 4) in aqueous media from iron salts, thiols, and sulfur, sulfide, or thiosulfate plus rhodonase. Inorg. Chem. 1985, 24, 4331–4335. [Google Scholar]
- Lo, W.; Scott, T.A.; Zhang, P.; Ling, C.C.; Holm, R.H. Stabilities of cubane type [Fe4S4(SR)4]2− clusters in partially aqueous media. J. Inorg. Biochem. 2011, 105, 497–508. [Google Scholar]
- Job, R.C.; Bruice, T.C. Iron-sulfur Clusters II: Kinetics of ligand exchange studied on a water-soluble Fe4S4(SR)4n− cluster. Proc. Natl. Acad. Sci. USA 1975, 72, 2478–2482. [Google Scholar]
- Dai, S.; Friemann, R.; Glauser, D.A.; Bourquin, F.; Manieri, W.; Schürmann, P.; Eklund, H. Structural snapshots along the reaction pathway of ferredoxin-thioredoxin reductase. Nature 2007, 448, 92–96. [Google Scholar]
- Welte, C.; Deppenmeier, U. Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta 2014, 1837, 1130–1147. [Google Scholar]
- Duin, E.C.; Madadi-Kahkesh, S.; Hedderich, R.; Clay, M.D.; Johnson, M.K. Heterodisulfide reductase from Methanothermobacter marburgensis contains an active-site [4Fe-4S] cluster that is directly involved in mediating heterodisulfide reduction. FEBS Lett. 2002, 512, 263–268. [Google Scholar]
- Shokes, J.E.; Duin, E.C.; Bauer, C.; Jaun, B.; Hedderich, R.; Koch, J.; Scott, R.A. Direct interaction of coenzyme M with the active-site Fe-S cluster of heterodisulfide reductase. FEBS Lett. 2005, 579, 1741–1744. [Google Scholar]
- Sakuraba, H.; Oshima, T. Novel energy metabolism in anaerobic hyperthermophilic archaea: A modified Embden-Meyerhof pathway. J. Biosci. Bioeng. 2002, 93, 441–448. [Google Scholar]
- Reher, M.; Gebhard, S.; Schonheit, P. Glyceraldehyde-3-phosphate ferredoxin oxidoreductase (GAPOR) and nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN), key enzymes of the respective modified Embden-Meyerhof pathways in the hyperthermophilic crenarchaeota Pyrobaculum aerophilum and Aeropyrum pernix. FEMS Microbiol. Lett. 2007, 273, 196–205. [Google Scholar]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl-sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. [Google Scholar]
- Rode, B.M. Peptides and the origin of life. Peptides 1999, 20, 773–786. [Google Scholar]
- Mulkidjanian, A.Y.; Galperin, M.Y.; Koonin, E.V. Co-evolution of primordial membranes and membrane proteins. Trends Biochem. Sci. 2009, 34, 206–215. [Google Scholar]
- Deamer, D.W. Proton permeation of lipid bilayers. J. Bioenerg. Biomembr. 1987, 19, 457–479. [Google Scholar]
- Hofvander, P.; Doan, T.T.; Hamberg, M. A prokaryotic acyl-CoA reductase performing reduction of fatty acyl-CoA to fatty alcohol. FEBS Lett. 2011, 585, 3538–3543. [Google Scholar]
- De Duve, C. Clues from present-day biology: The thioester world. In The Molecular Origins of Life; Brach, A., Ed.; Cambridge University Press: Cambridge, UK, 1998; pp. 219–236. [Google Scholar]
- Nivikov, Y.; Copley, S.D. Reactivity landscape of pyruvate under simulated hydrothermal vent conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 13283–13288. [Google Scholar]
- Wang, W.; Yang, B.; Qu, Y.; Liu, X.; Su, W. FeS/S/FeS2 redox system and its oxidoreductase-like chemistry in the iron-sulfur world. Astrobiology 2011, 11, 471–476. [Google Scholar]
- Fukuyama, T.; Lin, S.C.; Li, L. Facile reduction of ethyl thiol esters to aldehydes: Application to a total synthesis of a (+)-neothromycin A methyl ester. J. Am. Chem. Soc. 1990, 112, 7050–7051. [Google Scholar]
- Ragsdale, S.W. Pyruvate ferredoxin oxidoreductase and its radical intermediate. Chem. Rev. 2003, 103, 2333–2346. [Google Scholar]
- Chabrière, E.; Vernède, X.; Guigliarelli, B.; Charon, M.H.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystal structure of the free radical intermediate of pyruvate:ferredoxin oxidoreductase. Science 2001, 294, 2559–2563. [Google Scholar]
- Nakajima, T.; Yabushita, Y.; Tabushi, I. Amino acid synthesis through biogenetic-type CO2 fixation. Nature 1975, 256, 60–61. [Google Scholar]
- Tanaka, K.; Matsui, T.; Tanaka, T. Catalytic formation of α-keto acids by artifical CO2 fixation. J. Am. Chem. Soc. 1989, 111, 3765–3767. [Google Scholar]
- Olsen, R.K.; Currie, J.O. Synthetic uses of thiols. In The Chemistry of the Thiol Group, Part 2; Patai, S., Ed.; John Wiley & Sons: London, UK, 1974; pp. 519–588. [Google Scholar]
- Stacy, G.W.; Day, R.I.; Morath, R.J. Schiff bases and related substances II. Reactions of thiols with N-benzylidene aniline and N-benzylidene anthranilic acid. J. Am. Chem. Soc. 1955, 77, 3869–3873. [Google Scholar]
- Oakes, T.R.; Stacy, G.W. Reactions of thiols with Schiff bases in nonaqueous solvents. Addition equilibria, cleavage, and reduction. J. Am. Chem. Soc. 1972, 94, 1594–1600. [Google Scholar]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar]
- Kochi, H.; Kikuchi, G. Reactions of glycine synthesis and glycine cleavage catalyzed by extracts of Arthrobacter globiformis grown on glycine. Arch. Biochem. Biophys. 1969, 132, 359–369. [Google Scholar]
- Van Poelje, P.D.; Snell, E.E. Pyruvoyl-dependent enzymes. Annu. Rev. Biochem. 1990, 59, 29–59. [Google Scholar]
- Hess, U.; Theile, R. Electosynthesis of N-substituted dl-arylglycineesters and 1,2-diarylamino-1,2-diarylethanes by cathodic reduction of azomethines in the presence of carbon dioxide. J. Prakt. Chem. 1982, 324, 385–399. [Google Scholar]
- Silvestri, G.; Gambino, S.; Filardo, G. Electrochemical syntheses involving carbon dioxide. In Enzymatic and Model Carboxylation and Reduction Reactions for Carbon Dioxide Utilization; Arresta, M., Schloss, M.V., Eds.; Kluwer Academic Publishers: South Holland, The Netherlands, 1990; pp. 101–127. [Google Scholar]
- Nakada, H.I.; Weinhouse, S. Non-enzymatic transamination with glyoxylic acid and various amino acids. J. Biol. Chem. 1953, 204, 831–836. [Google Scholar]
- Howard, B.R.; Endrizzi, J.A.; Remington, S.J. Crystal structure of Escherichia coli malate synthase G complexed with magnesium and glyoxylate at 2.0 Å resolution: Mechanistic implications. Biochemistry 2000, 39, 3156–3168. [Google Scholar]
- Hsu, R.Y.; Mildvan, A.; Chang, G.; Fung, C. Mechanism of malic enzyme from pigeon liver. Magnetic resonance and kinetic studies of the role of Mn2+. J. Biol. Chem. 1976, 251, 6574–6583. [Google Scholar]
- Aktas, D.F.; Cook, P.F. A lysine-tyrosine pair carries out acid-base chemistry in the metal ion-dependent pyridine dinucleotide-linked β-hydroxyacid oxidative decarboxylases. Biochemistry 2009, 48, 3565–3577. [Google Scholar]
- Goward, C.R.; Nicholls, D.J. Malate dehydrogenase: A model for structure, evolution, and catalysis. Protein Sci. 1994, 3, 1883–1888. [Google Scholar]
- Mukhopadhyay, B.; Stoddard, S.F.; Wolfe, R.S. Purification, regulation, and molecular and biochemical characterization of pyruvate carboxylase from Methanobacterium thermoautotrophicum strain ΔH. J. Biol. Chem. 1998, 273, 5155–5166. [Google Scholar]
- Flint, D.H. Escherichia coli fumarase A catalyzes the isomerization of enol and keto oxalacetic acid. Biochemistry 1993, 32, 799–805. [Google Scholar]
- White, R.H. Intermediates in the biosynthesis of Coenzyme M (2-mercaptoethane sulfonic acid). Biochemistry 1986, 25, 5304–5308. [Google Scholar]
- White, R.H. Biosynthesis of the 7-mercaptoheptanoic acid subunit of component B [(7-mercaptoheptanoyl) threonine phosphate] of methanogenic bacteria. Biochemistry 1989, 28, 860–865. [Google Scholar]
- Dörr, M.; Käßbohrer, J.; Grunert, R.; Kreisel, G.; Brand, W.A.; Werner, R.A.; Geilmann, H.; Apfel, C.; Robl, C.; Weigand, W.; et al. A possible prebiotic formation of ammonia from dinitrogen on iron sulfide surfaces. Angew. Chem. Int. Ed. 2003, 42, 1540–1543. [Google Scholar]
- Robertson, M.P.; Joyce, G.F. The origins of the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Braud, C.; Brunel, C.; Vert, M. Poly(β-malic acid): A new polymeric drug-carrier. Polym. Bull. (Berl.) 1985, 13, 293–299. [Google Scholar]
- Karl, M.; Anderson, R.; Holler, E. Injection of poly(β-l-malate) into the plasmodium of Physarum polycephalum shortens the cell cycle and increases the growth rate. Eur. J. Biochem. 2004, 271, 3805–3811. [Google Scholar]
- Liu, S.; Steinbüchel, A. Investigation of poly(β-l-malic acid) production by strains of Aureobasidium pullulans. Appl. Microbiol. Biotechnol. 1996, 46, 273–278. [Google Scholar]
- Goodman, I. Polyesters, Volume I: Saturated Polymers; Elsevier: New York, NY, USA, 1965. [Google Scholar]
- Kasting, J.F. Earth’s earliest atmosphere. Science 1993, 259, 920–926. [Google Scholar]
- Portilla-Arias, J.A.; García-Alvarez, M.; Martínez de Ilarduya, A.; Holler, E.; Muñoz-Guerra, S. Thermal decomposition of fungal poly(β-l-malic acid) and poly(β-l-malate)s. Biomacromolecules 2006, 7, 3283–3290. [Google Scholar]
- Van der Sluis, P.; Kroon, J. The structure of (+/−) malic acid, (+/−)-C4H6O5. Acta Crystallogr. 1985, C41, 956–959. [Google Scholar]
- Gavezzotti, A. Hydrogen bond strength and bond geometry in cyclic dimers of crystalline carboxylic acids. Acta Crystallogr. B 2008, 864, 401–403. [Google Scholar]
- Heisler, I.A.; Mazur, K.; Yamaguchi, S.; Tominaga, K.; Meech, S.R. Measuring acetic acid dimer modes by ultra fast time-domain Raman spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 15573–15579. [Google Scholar]
- Losada, M.; Tran, H.; Xu, Y. Lactic acid in solution: Investigations of lactic acid self-aggregation and hydrogen bonding interactions with water and methanol using vibrational absorption and vibrational circular dichroism spectroscopies. J. Chem. Phys. 2008, 128. [Google Scholar] [CrossRef]
- Nakamura, A.; Yao, M.; Chimnaronk, S.; Sakai, N.; Tanaka, I. Ammonia channel couples glutaminase with transamidase reactions in GatCAB. Science 2006, 312, 1954–1958. [Google Scholar]
- Beccera-Figueroa, L.; Ojeda-Porras, A.; Gamba-Sánchez, D. Transamidation of carboxamides catalyzed by Fe(III) and water. J. Org. Chem. 2014, 79, 4544–4552. [Google Scholar]
- Levy, M.; Silberman, D.E. The reactions of amino and imino acids with formaldehyde. J. Biol. Chem. 1937, 118, 723–734. [Google Scholar]
- Kitamoto, Y.; Maeda, H. Reevaluation of the reaction of formaldehyde at low concentrations with amino acids. J. Biochem. 1980, 87, 1519–1530. [Google Scholar]
- Argyrou, A.; Washabaugh, M.W.; Pickart, C.M. Dihydroorotate dehydrogenase from Clostridium oroticum is a class 1B enzyme and utilizes a concerted mechanism of catalysis. Biochemistry 2000, 39, 10373–10384. [Google Scholar]
- Wise, E.; Yew, W.S.; Babbitt, P.C.; Gerlt, J.A.; Rayment, I. Homologous (β/α)8-barrel enzymes that catalyze unrelated reactions: Orotidine-5'-monophosphate decarboxylase and 3-keto-l-gulonate-6-phosphate decarboxylase. Biochemistry 2002, 41, 3861–3869. [Google Scholar]
- Mittapalli, G.K.; Osornio, Y.M.; Guerrero, M.A.; Reddy, K.R.; Krishnamurthy, R.; Eschenmoser, A. Mapping the landscape of potentially primordial informational oligomers: Oligodipeptides tagged with 2,4-disubstituted 5-aminopyrimidines as recognition elements. Angew. Chem. Int. Ed. 2007, 46, 2478–2484. [Google Scholar]
- Stuecker, T.N.; Hodge, K.M.; Escalante-Semerena, J.C. The missing link in coenzyme A biosynthesis: PanM (formerly YhhK), a yeast GCN5 acetyltransferase homologue triggers aspartate decarboxylase (PanD) maturation in Salmonella enterica. Mol. Microbiol. 2012, 84, 608–619. [Google Scholar]
- Chladek, S. The synthesis, reactions and properties of the 2'(3')-O-aminoacyl and peptidyl nucleosides and nucleotides. In Chemistry of Nucleosides and Nucleotides; Townsend, L.B., Ed.; Springer Science & Business Media: New York, NY, USA, 1994; Volume 3, pp. 107–143. [Google Scholar]
- Gavrilova, L.P.; Kostiashkina, O.E.; Koteliansky, V.E.; Rutkevitch, N.M.; Spirin, A.S. Factor-free (“non-enzymic”) and factor-dependent systems of translation of polyuridylic acid by Escherichia coli ribosomes. J. Mol. Biol. 1976, 101, 537–552. [Google Scholar]
- Southworth, D.R.; Brunelle, J.L.; Green, R. EFG-independent translocation of the mRNA:tRNA complex is promoted by modification of the ribosome with thiol-specific reagents. J. Mol. Biol. 2002, 324, 611–623. [Google Scholar]
- Rich, A. On the problems of evolution and biochemical information transfer. In Horizons in Biochemistry; Kasha, M., Pullman, B., Eds.; Academic Press: New York, NY, USA, 1962; pp. 103–126. [Google Scholar]
- Weber, A.L.; Orgel, L.E. Poly(U)-directed peptide bond formation from the 2'(3')-glycyl esters of adenosine derivatives. J. Mol. Evol. 1980, 16, 1–10. [Google Scholar]
- Reusch, R.N. Physiological importance of poly-(R)-3-hydroxybutyrates. Chem. Biodivers. 2012, 9, 2343–2366. [Google Scholar]
- Brooks, D.J.; Fresco, J.R. Increased frequency of cysteine, tyrosine, and phenylalanine residues since the last universal ancestor. Mol. Cell. Proteomics 2002, 1, 125–131. [Google Scholar]
- Francis, B.R. Evolution of the genetic code by incorporation of amino acids that improved or changed protein function. J. Mol. Evol. 2013, 77, 134–158. [Google Scholar]
- Eigen, M.; Winkler-Oswatitsch, R. Transfer-RNA, an early gene? Naturwissenschaften 1981, 68, 217–228. [Google Scholar]
- Ikehara, K.; Omori, Y.; Arai, R.; Hirose, A. A novel theory on the origin of the genetic code: A GNC-SNS hypothesis. J. Mol. Evol. 2002, 54, 530–538. [Google Scholar]
- Higgs, P.G. A four-column theory for the origin of the genetic code: Tracing the evolutionary pathways that gave rise to an optimized code. Biol. Direct 2009, 4. [Google Scholar] [CrossRef]
- Van der Gulik, P.; Massar, S.; Gilis, D.; Burhman, H.; Rooman, M. The first peptides: The evolutionary transition between prebiotic amino acids and early proteins. J. Theor. Biol. 2009, 261, 531–539. [Google Scholar]
- Fujihashi, M.; Numoto, N.; Kobayashi, Y.; Mizushima, A.; Tsujimura, M.; Nakamura, A.; Kawarabayasi, Y.; Miki, K. Crystal structure of archaeal photolyase from Sulfolobus tokodaii with two FAD molecules: Implication of a novel light-harvesting cofactor. J. Mol. Biol. 2007, 365, 903–910. [Google Scholar]
- Deamer, D.W.; Dworkin, J.P.; Sandford, S.A.; Bernstein, M.P.; Allamandola, L.J. The first cell membranes. Astrobiology 2002, 2, 371–381. [Google Scholar]
- Segré, D.; Ben-Eli, D.; Deamer, D.W.; Lancet, D. The Lipid World. Orig. Life Evol. Biosph. 2001, 31, 119–145. [Google Scholar]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. Lond. 1997, 154, 377–402. [Google Scholar]
- Alpermann, T.; Rüdel, K.; Rüger, R.; Steiniger, F.; Nietzsche, S.; Filiz, V.; Förster, S.; Fahr, A.; Weigand, W. Polymersomes containing iron sulfide (FeS) as primordial cell model: For the investigation of energy providing redox reactions. Orig. Life Evol. Biosph. 2010, 41, 103–119. [Google Scholar]
- Menzel, K.; Menzel, K.; Apfel, U.P.; Wolter, N.; Rüger, R.; Alpermann, T.; Steiniger, F.; Gabel, D.; Förster, S.; Weigand, W.; et al. [FeFe]-hydrogenase models assembled into vesicular structures. J. Liposome Res. 2014, 24, 59–68. [Google Scholar]
- Summers, D.P.; Noveron, J.; Basa, R.C. Energy transduction inside of amphiphilic vesicles: Encapsulation of photochemically active semiconducting particles. Orig. Life Evol. Biosph. 2009, 39, 127–140. [Google Scholar]
- Emiliozzi, R.; Pichat, L.; Herbert, M. Preparation of cysteine-S35 hydrochloride. Bull. Soc. Chem. Fr. 1959, 1544–1545. [Google Scholar]
- Orgel, L.E. Some consequences of the RNA world hypothesis. Orig. Life Evol. Biosph. 2003, 33, 211–218. [Google Scholar]
- Ross, D.S. The viability of a nonenzymatic reductive citric acid cycle—Kinetics and thermochemistry. Orig. Life Evol. Biosph. 2007, 37, 61–65. [Google Scholar]
- Orgel, L.E. The implausibility of metabolic cycles on the prebiotic Earth. PLoS Biol. 2008, 6. [Google Scholar] [CrossRef]
- Copley, S.D.; Smith, E.; Morowitz, H.J. The emergence of sparse metabolic networks. J. Cosmol. 2010, 10, 3345–3361. [Google Scholar]
- Engelhart, A.E.; Hud, N.V. Primitive genetic polymers. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Szathmáry, E. Why are there four letters in the genetic alphabet? Nat. Rev. Genet. 2003, 4, 995–1001. [Google Scholar]
- Calvin, M. Chemical Evolution: Molecular Evolution towards the Origin of Living Systems on the Earth and Elsewhere; Clarendon Press: London, UK, 1969. [Google Scholar]
- Rauchfuss, H. Chemical Evolution and the Origin of Life; Springer-Verlag: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Schrum, J.P.; Zhu, T.F.; Szostak, J.W. The origins of cellular life. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Mansy, S.S.; Schrum, J.P.; Krishnamurthy, M.; Tobé, S.; Treco, D.A.; Szostak, J.W. Template-directed synthesis of a genetic polymer in a model protocell. Nature 2008, 454, 122–125. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, B.R. The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells. Life 2015, 5, 467-505. https://doi.org/10.3390/life5010467
Francis BR. The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells. Life. 2015; 5(1):467-505. https://doi.org/10.3390/life5010467
Chicago/Turabian StyleFrancis, Brian R. 2015. "The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells" Life 5, no. 1: 467-505. https://doi.org/10.3390/life5010467
APA StyleFrancis, B. R. (2015). The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells. Life, 5(1), 467-505. https://doi.org/10.3390/life5010467