
Genetic Code Evolution Reveals the Neutral Emergence of Mutational Robustness, and Information as an Evolutionary Constraint

Abstract
:1. The Genetic Code: Near Optimal and Near Universal
2. Neutral Emergence of Error Minimization in the Genetic Code
2.1. The Non-Adaptive Code Hypothesis


2.2. Emergence in Biological Systems


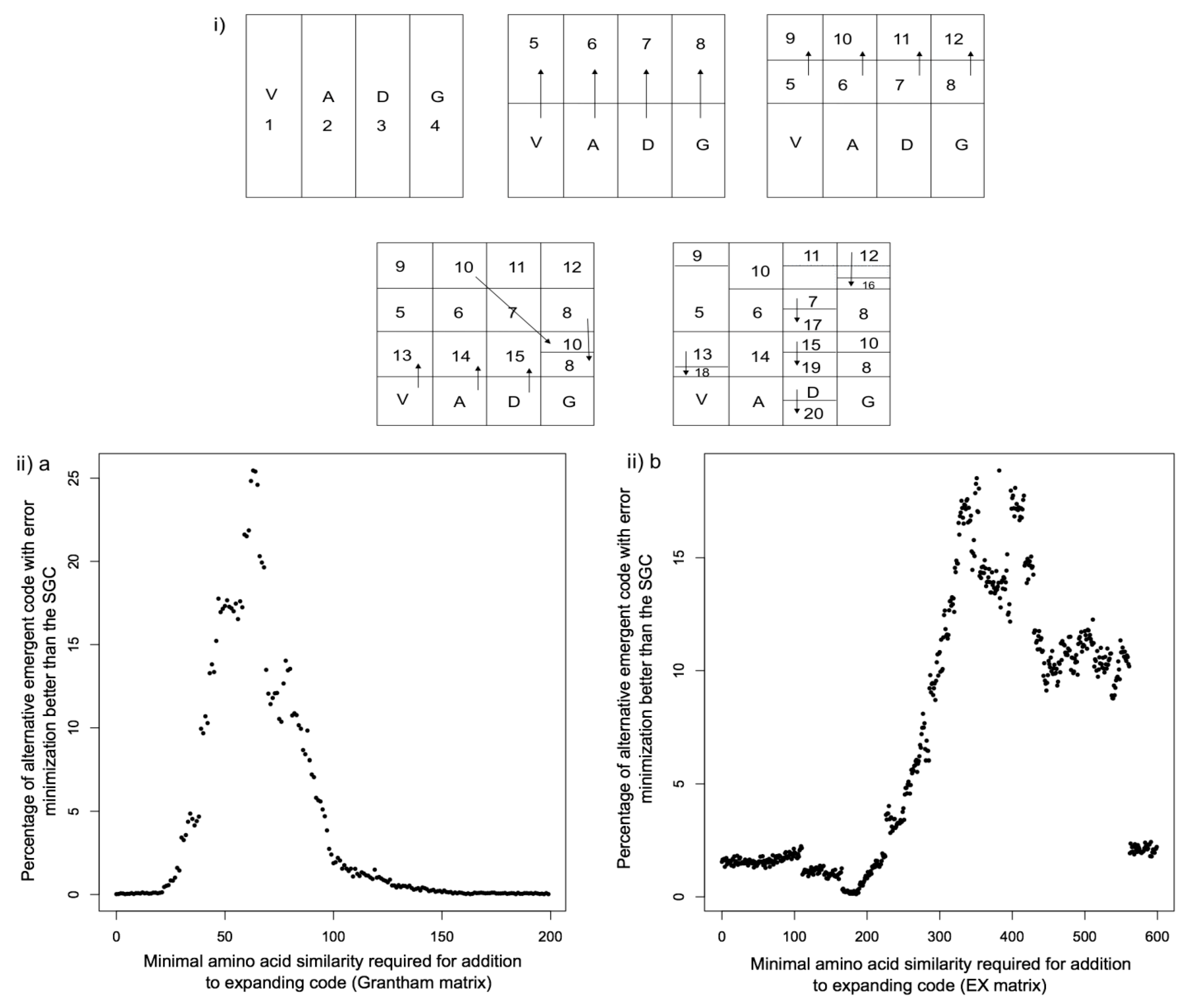{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Potential driving force | Indirect benefit (neutrally emergent*) |
|---|---|---|
| Increased proteome hydrophobicity in AT rich genomes | Hypothesized that AT bias may arise neutrally via changes in mutation bias [83], one cause of which may be loss of DNA repair genes [84], which may indirectly be a result of a reduction in P [85,86,87], and this work. AT rich codons encode more hydrophobic amino acids, so AT bias results in more hydrophobic proteins | Increased hydrophobicity of proteome results in increased protein folding stability [88] * |
| Scale free structure of metabolic networks | There is evidence preferential attachment has given rise to the scale free property [89] | Robustness to gene deletion [90,91] * |
| Scale free structure of protein interaction networks | There is evidence that preferential attachment has given rise to the scale free property [92,93] | Robustness to gene deletion [94,95] * |
| Scale free structure of gene regulatory networks | There is evidence a combination of gene duplication and preferential attachment are responsible for the scale free property [96] | Robustness to mutation [97] * |
| Survival of the flattest | Survival of the flattest refers to the increase in number of robust organisms in a population when mutation rates are high. This neutrally emerges in digital organisms [98] and RNA viruses [40] in the absence of direct selection for the property | Increased robustness of the population to mutation * |
| Mutational robustness of protein and RNA structures | Mutational robustness in RNA secondary structures [74,99], protein 2D lattices [73,100] and 3D coarse grained protein models [29] neutrally emerges via random movement on a neutral network as a result of genetic drift | Increased structural robustness to mutation * |
| Error minimization of the genetic code | There is evidence that error minimization neutrally emerged during genetic code expansion via gene duplication of adaptor molecules and charging enzymes [21] and this work. | Error minimization reduces the deleterious impact of point mutations, transcriptional and translational errors * |
| Genetic dominance | It has been proposed that genetic dominance is selected for to increase metabolic flux [101], or that it is a side product of enzyme kinetics [102] | Increased mutational robustness [38] |
| Enhanced DNA repair in Deinococcus radiodurans | The ability to withstand dessication may have led to the enhanced repair of double stranded breaks [103] | Enhanced repair of double stranded breaks also leads to radiation resistance in this species. Radiation is rarely encountered in nature, so it is unlikely radiation resistance was directly selected for [103] |
| Trait | Potential driving force | Indirect benefit (neutrally emergent *) |
|---|---|---|
| Sexual reproduction | The purpose of sexual reproduction has been proposed to be DNA repair via recombination [104] | Recombination leads to a reduction in the Hill-Robertson effect, enhancing the strength of selection * |
| Segmentation of virus genomes | The role of virus genome segmentation has been linked to differential gene expression [105] | In cystoviruses, segmentation leads to random assortment, and subsequent amelioration of linkage disequilibrium [106], increasing the power of selection. Likewise, in the influenza virus segmentation may increase the strength of selection [107] * |
| Protein domain shuffling | Domain shuffling is facilitated by the occurrence of introns [108], which have a variety of functions, however the role of most of them remains to be established [109] | Domain shuffling has been linked to evolutionary innovation [110] * |
| Reduced population size | Many factors may act to reduce population size and it is unlikely to be directly selected for | Ability to traverse evolutionary barriers [111,112] * |
| Nonfunctional DNA in higher eukaryotes | The function of the majority of intron sequences and intergenic DNA, if any, has not been established. Notably, overall there is a lack of sequence conservation, indicating a lack of sequence specific selection [113] | Longer introns and intergenic DNA regions lead to an increase in recombination events, reducing the Hill-Roberston effect and so increasing the strength of selection [114,115,116] * |
| Evolutionary capacitance of HSP90 | HSP90 is a normal part of the stress response in the eukaryotes | HSP90 acts to store cryptic genetic variation, this is exposed in times of stress due to a reduction in the concentration of free HSP90 [117,118] * |
| Evolutionary capacitance of complex gene regulatory networks | Gene regulatory network structure is driven by the addition and removal of nodes, according to the immediate selective benefit | The loss of a gene enhances the phenotypic variation of remaining components of the network, and this promotes evolvability, this effect is not dependent on network topology [119] * |
| Error minimization in the SGC | There is evidence that error minimization has neutrally emerged as a consequence of genetic code expansion over time [21,30], and this work | Error minimization has been proposed to result in the increased evolvability of proteins [78,79] * |
| Elevated mutation rates in RNA viruses | The ultimate cause of elevated mutation rates in RNA viruses has not established, but reduced P may be a factor [85] and this work. The proximate cause of the elevated mutation rates is a lack of proofreading in the replicative polymerase | Elevated mutation rates increase the ability to evade the host immune system and adapt to drug treatments |
| Ambiguous decoding of the CUG codon as both serine and leucine in Candida yeasts | The ambiguous decoding of CUG [120] appears to have been a factor in the codon reassignment of CUG leu→ser [120] | Ambiguous CUG decoding produces elevated levels of HSPs and this enhances survivability in challenging environments [121] |
| Polyploidy | Polyploidy is caused by abnormal cell division | Polyploidy is proposed to result in increased evolvability in plants [122,123] |
| Lateral gene transfer (LGT) in prokaryotes | LGT may have a role in DNA repair of the prokaryotic genome [124] or may be a side-product of the uptake of DNA as carbon and energy source [125] | LGT leads to increased evolvability in response to environmental challenges |
2.3. Pseudaptations: Beneficial Traits that Have not Been Directly Selected for
3. Proteome Size as a Constraint on the Genetic Code
3.1. Unfreezing of the Code
| Lineage and phylogenetic affiliation | Genetic code change | Genome size | Genome GC content | Elevated substitution rate? | Loss of DNA repair? | Habitat |
|---|---|---|---|---|---|---|
| Mycoplasmas (Mollicutes) | UGA (stop)→trp [131] | 580–1359 kbp (Genbank) | 25%–40% (Genbank) | Yes [132] | Yes [133] | Vertebrate cells |
| Spiroplasmas (Mollicutes) | UGA (stop)→trp [134] | 940–2220 kbp [135] | 29% [ 136] | Yes [132] | Yes [137] | Insect and plant cells |
| Ureaplasmas (Mollicutes) | UGA (stop)→trp [138] | 750–950 kbp [139] | 25% [139] | Yes [132] | Not determined | Vertebrate cells |
| SR1 bacteria (related to Chloroflexi) | UGA (stop)→gly [140] | 1178 kbp [141] | 31% [141] | Yes [141] | Not determined | Human body (extracellular), sediments |
| Nasuia deltocephalinicol (β proteobacteria) | UGA (stop)→trp [142] | 112 kbp [142] | 17% [142] | Yes [142] | Yes [142] | Circada (insect) cells |
| Sulcia muelleri (Bacteroidetes) | UGA (stop)→trp [142] | 190 kbp [142] | 24% [142] | Yes [143] | Yes [143] | Sharpshooter (insect) cells |
| Hodgkinia cicadicola (α proteobacteria) | UGA→trp [144] | 144 kbp [144] | 58% [144] | Yes [144] | Yes [144] | Circada (insect) cells |
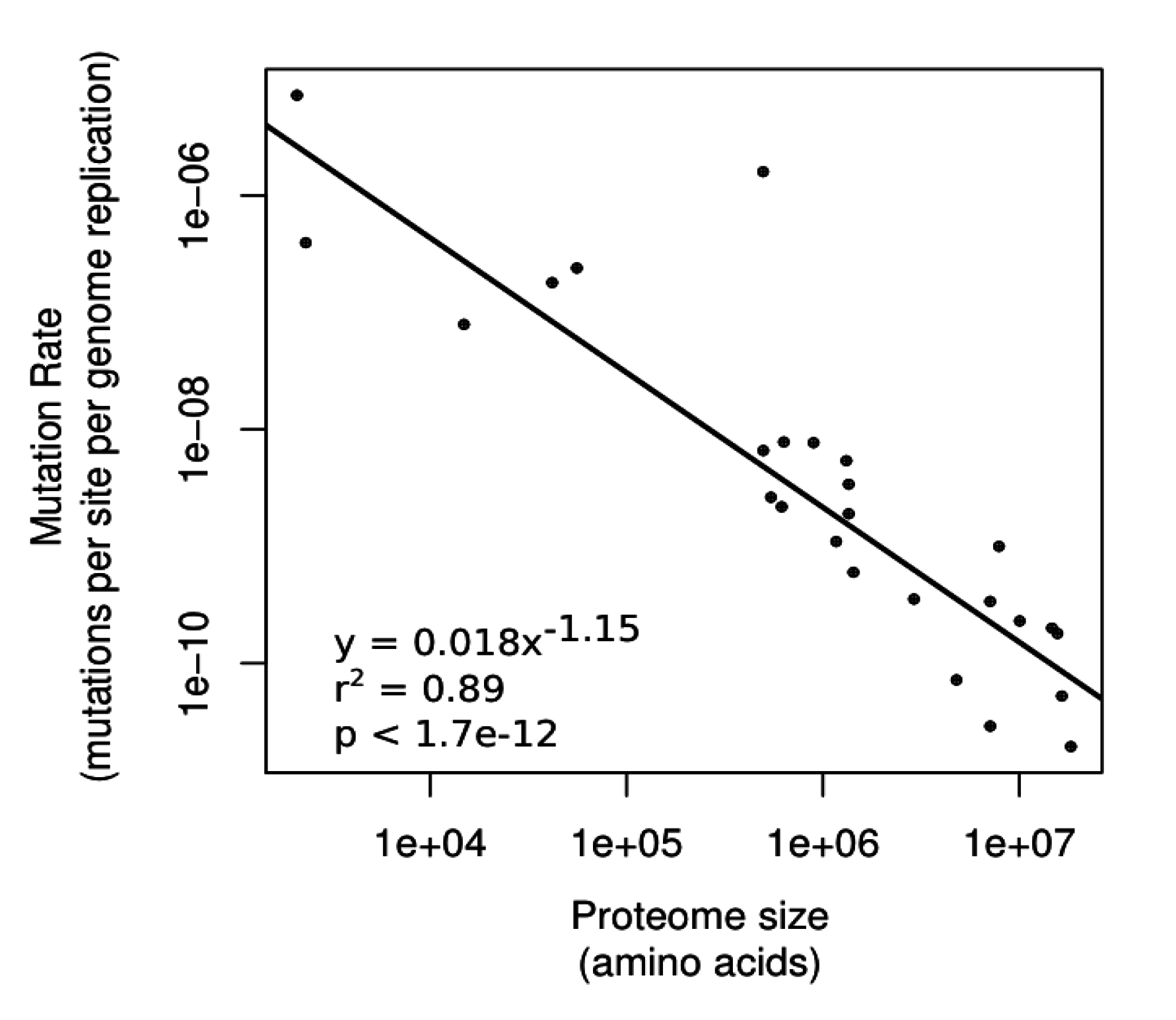
3.2. Genomic Information Content as a Constraint on Genetic Fidelity
3.2.1. Differences in Underlying Mutation Rates
3.2.2. Loss of DNA Repair Genes and Changes in Genome GC Content

3.2.3. The Evolution of Sexual Reproduction
3.2.4. Inefficient Organelle Protein Translation
3.3. Information as a Constraint in Diverse Systems
| Discipline | Parameter |
|---|---|
| Information theory | Shannon entropy/message length |
| Signalling games | Complete/incomplete/perfect information |
| Physics | Physical information |
| Economics | Information goods |
| Linguistics | Word/sentence length is related to information content |
| Ecology | Alpha diversity |
| Complexity theory | Complexity measures are related to information content |
| Biology | Genomic information content, organismal complexity |
| System | Nature of informational/complexity constraint | Consequence |
|---|---|---|
| Business | Complexity of business | “Complexity costs“ add financial burden on the business |
| Healthcare | Complexity of medical treatments | Increased probability of error and consequent detrimental health outcomes [191] |
| Statistical models | Number of parameters in a model | Greater number of parameters increases the variance of outcome [192] |
| Messages in communication systems | Message length | Greater message length in communications is costly, leading to the noiseless coding theorum which formalizes message compression [185] |
| Computer programming | Complexity of code, “feature creep” | Increased production costs |
| Ecosystem | Biodiversity/number of endemic species | The more biodiverse an ecosystem, the greater the political/economic pressure to preserve it |
| Biological research | Equation density in a research paper | Reduced citation of paper [193] |
| Genomics | Quantity and complexity of high throughput data | Analysis costs, i.e., the “bioinformatics bottleneck” |
| Multicellular animals | Body size | More cells (and so genome copies) proposed to increase cancer risk [194,195,196] |
| Lateral gene transfer | Complexity of protein complexes | The complexity hypothesis proposes that participation in multi-subunit protein complexes constitutes a barrier to the lateral transfer of informational genes [197] |
| Organismal evolution | Organismal complexity | Organismal complexity proposed to constrain rate of adaptation [175,198] |
| Molecular evolution | Genomic information content | Proposed to constrain genetic fidelity [85,86,87,161,164,165] and this work |
4. Conclusions
Acknowledgements
Supplementary Materials
Conflicts of Interest
References
- Koonin, E.V.; Novozhilov, A.S. Origin and evolution of the genetic code: The universal enigma. IUBMB Life 2009, 61, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Sonneborn, T.M. Degeneracy of the genetic code: Extent, nature, and genetic implications. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Woese, C.R. On the evolution of the genetic code. Proc. Natl. Acad. Sci. USA 1965, 54, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.J. Role of the amino-acid “code” and of selection for conformation in the evolution of proteins. Nature 1966, 210, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Wittes, R.E. Genetic Code: Aspects of Organization. Science 1966, 153, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J.; Hurst, L.D. The Genetic Code Is One in a Million. J. Mol. Evol. 1998, 47, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J.; Knight, R.D.; Landweber, L.F.; Hurst, L.D. Early Fixation of an Optimal Genetic Code. Mol. Biol. Evol. 2000, 17, 511–518. [Google Scholar] [CrossRef]
- Gilis, D.; Massar, S.; Cerf, N.J.; Rooman, M. Optimality of the genetic code with respect to protein stability and amino-acid frequencies. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Goodarzi, H.; Nejad, H.A.; Torabi, N. On the optimality of the genetic code, with the consideration of termination codons. Biosystems 2004, 77, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Goldenfeld, N.; Mathew, D.; Luthey-Schulten, Z. Extreme genetic code optimality from a molecular dynamics calculation of amino acid polar requirement. Phys. Rev. E 2009, 79, 060901. [Google Scholar] [CrossRef]
- Buhrman, H.; van der Gulik, P.T.; Klau, G.W.; Schaffner, C.; Speijer, D.; Stougie, L. A realistic model under which the genetic code is optimal. J. Mol. Evol. 2013, 77, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Alff-Steinberger, C. The Genetic Code and Error Transmission. Proc. Natl. Acad. Sci. USA 1969, 64, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.; Wu, T.; Keulmann, N. The Case for an Error Minimizing Standard Genetic Code. Orig. Life Evol. Biosph. 2003, 33, 457–477. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Role of minimization of chemical distances between amino acids in the evolution of the genetic code. Proc. Natl. Acad. Sci. USA 1980, 77, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. The extension reached by the minimization of the polarity distances during the evolution of the genetic code. J. Mol. Evol. 1989, 29, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N. Further results on error minimization in the genetic code. J. Mol. Evol. 1993, 37, 662–664. [Google Scholar] [PubMed]
- Judson, O.P.; Haydon, D. The Genetic Code: What Is It Good For? An Analysis of the Effects of Selection Pressures on Genetic Codes. J. Mol. Evol. 1999, 49, 539–550. [Google Scholar]
- Di Giulio, M.; Medugno, M. The Level and Landscape of Optimization in the Origin of the Genetic Code. J. Mol. Evol. 2001, 52, 372–382. [Google Scholar] [PubMed]
- Novozhilov, A.; Wolf, Y.; Koonin, E. Evolution of the genetic code: partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol. Direct 2007, 2. [Google Scholar] [CrossRef]
- Massey, S.E. A Neutral Origin for Error Minimization in the Genetic Code. J. Mol. Evol. 2008, 67, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. The Origin of the Genetic Code cannot be Studied using Measurements based on the PAM Matrix because this Matrix Reflects the Code Itself, Making any such Analyses Tautologous. J. Theor. Biol. 2001, 208, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Najafabadi, H.S.; Hassani, K.; Nejad, H.A.; Torabi, N. On the optimality of the genetic code, with the consideration of coevolution theory by comparison of prominent cost measure matrices. J. Theor. Biol. 2005, 235, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Dugre, D.H.; Saxinger, W.C.; Dugre, S.A. The molecular basis for the genetic code. Proc. Natl. Acad. Sci. USA 1966, 55, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar]
- Crick, F.H. The origin of the genetic code. J Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Lewontin, R.C. The spandrels of San Marco and the Panglossian paradigm: A critique of the adaptionist programme. Proc. R. Soc. Lond. B 1979, 205, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. Pseudaptations and the Emergence of Beneficial Traits. In Evolutionary Biology—Concepts, Molecular and Morphological Evolution; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 81–98. [Google Scholar]
- Pagan, R.; Massey, S.E. A Nonadaptive Origin of a Beneficial Trait: In Silico Selection for Free Energy of Folding Leads to the Neutral Emergence of Mutational Robustness in Single Domain Proteins. J. Mol. Evol. 2014, 78, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. Searching of Code Space for an Error-Minimized Genetic Code Via Codon Capture Leads to Failure, or Requires At Least 20 Improving Codon Reassignments via the Ambiguous Intermediate Mechanism. J. Mol. Evol. 2010, 70, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Jukes, T.H. Evolution of the genetic code as affected by anticodon content. Trends Genet. 1988, 4, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Jukes, T. Codon reassignment (codon capture) in evolution. J. Mol. Evol. 1989, 28, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.W.; Yarus, M. Transfer RNA Mutation and the Malleability of the Genetic Code. J. Mol. Biol. 1994, 235, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.; Yarus, M. On malleability in the genetic code. J. Mol. Evol. 1996, 42, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Andachi, Y.; Muto, A.; Osawa, S. CGG: An unassigned or nonsense codon in Mycoplasma capricolum. Proc. Natl. Acad. Sci. USA 1991, 88, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Kano, A.; Andachi, Y.; Ohama, T.; Osawa, S. Novel anticodon composition of transfer RNAs in Micrococcus luteus, a bacterium with a high genomic G+C content: Correlation with codon usage. J. Mol. Biol. 1991, 221, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E.; Garey, J. A Comparative Genomics Analysis of Codon Reassignments Reveals a Link with Mitochondrial Proteome Size and a Mechanism of Genetic Code Change via Suppressor tRNAs. J. Mol. Evol. 2007, 64, 399–410. [Google Scholar] [CrossRef] [PubMed]
- De Visser, J.A.G.M.; Hermisson, J.; Wagner, G.P.; Meyers, L.A.; Bagheri-Chaichian, H.; Blanchard, J.L.; Chao, L.; Cheverud, J.M.; Elena, S.F.; Fontana, W.; et al. Perspective: Evolution and Detection of Genetic Robustness. Evolution 2003, 57, 1959–1972. [Google Scholar]
- Elena, S.F.; Carrasco, P.; Daròs, J.-A.; Sanjuán, R. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 2006, 7, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Cuevas, J.M.; Furió, V.; Holmes, E.C.; Moya, A. Selection for Robustness in Mutagenized RNA Viruses. PLoS Genet. 2007, 3, e93. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Willendorfer, M.; Nowak, M.A. Why are phenotypic mutation rates much higher than genotypic mutation rates? Genetics 2006, 172, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Selection on codon usage for error minimization at the protein level. J. Mol. Evol. 2004, 59, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Lehmann, J.; Omidi, M. Error minimization explains the codon usage of highly expressed genes in Escherichia coli. Gene 2007, 387, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: Selection for translational accuracy. J. Mol. Evol. 2007, 24, 374–381. [Google Scholar] [CrossRef]
- Cusack, B.P.; Arndt, P.F.; Duret, L.; Crollius, H.R. Preventing dangerous nonsense: Selection for robustness to transcriptional error in human genes. PLoS Genet. 2011, 7, e1002276. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, T.; Kurnaz, I.A.; Wagner, A. Selection shapes the robustness of ligand-binding amino acids. J. Mol. Evol. 2013, 76, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, R.; Smit, S.; Knight, R. Do universal codon-usage patterns minimize the effects of mutation and translation error? Genome Biol. 2005, 6. [Google Scholar] [CrossRef]
- Zhu, C.-T.; Zeng, X.-B.; Huang, W.-D. Codon usage decreases the error minimization within the genetic code. J. Mol. Evol. 2003, 57, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Codon usage bias and mutation constraints reduce the level of error minimization of the genetic code. J. Mol. Evol. 2004, 59, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Woese, C. The Genetic Code: The Molecular Basis for Genetic Expression; Harper and Row: New York, NY, USA, 1967. [Google Scholar]
- Morgens, D.; Cavalcanti, A.O. An Alternative Look at Code Evolution: Using Non-canonical Codes to Evaluate Adaptive and Historic Models for the Origin of the Genetic Code. J. Mol. Evol. 2013, 76, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kurnaz, M.; Bilgin, T.; Kurnaz, I. Certain Non-Standard Coding Tables Appear to be More Robust to Error than the Standard Genetic Code. J. Mol. Evol. 2010, 70, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Stoltzfus, A.; Yampolsky, L.Y. Amino acid exchangeability and the adaptive code hypothesis. J. Mol. Evol. 2007, 65, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Illangasekare, M.; Sanchez, G.; Nickles, T.; Yarus, M. Aminoacyl-RNA synthesis catalyzed by an RNA. Science 1995, 267, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Bessho, Y.; Wei, K.; Szostak, J.W.; Suga, H. Ribozyme-catalyzed tRNA aminoacylation. Nat. Struct. Biol. 2000, 7, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Yampolsky, L.Y.; Stoltzfus, A. The exchangeability of amino acids in proteins. Genetics 2005, 170, 1459–1472. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. A Sequential “2–1–3” Model of Genetic Code Evolution That Explains Codon Constraints. J. Mol. Evol. 2006, 62, 809–810. [Google Scholar] [CrossRef] [PubMed]
- Gamow, G. Possible relation between deoxyribonucleic acid and protein structures. Nature 1954, 173. [Google Scholar] [CrossRef]
- Dunnill, P. Triplet nucleotide-amino-acid pairing; a stereochemical basis for the division between protein and non-protein aminoacids. Nature 1966, 210, 1267–1268. [Google Scholar] [CrossRef]
- Pelc, S.R.; Welton, M.G.E. Stereochemical relationship between coding triplets and amino-acids. Nature 1966, 209, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. A co-evolution theory of the genetic code. Proc. Natl. Acad. Sci. USA 1975, 72, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. The evolution of a universal genetic code. Proc. Natl. Acad. Sci. USA 1976, 73, 2336–2340. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T. Coevolution theory of the genetic code at age thirty. BioEssays 2005, 27, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Tong, K.; Marck, C.; Grosjean, H.; Wong, J.T. Transfer RNA paralogs: Evidence for genetic code-amino acid biosynthesis coevolution and an archaeal root of life. Gene 2003, 310, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.S. A System of Logic, Ratiocinative and Inductive: Being a Connected View of the Principles of Evidence and the Methods of Scientific Investigation; John W. Parker: London, UK, 1843. [Google Scholar]
- Weibel, E.R. Fractal geometry: A design principle for living organisms. Am. J. Physiol. 1991, 261, 361–369. [Google Scholar]
- Leisman, G.; Koch, P. Networks of conscious experience: computational neuroscience in understanding life, death and consciousness. Rev. Neurosci. 2009, 20, 151–176. [Google Scholar] [PubMed]
- Albert, R.; Jeong, H.; Barabasi, A. Error and attack tolerance of complex networks. Science 2000, 406, 378–382. [Google Scholar]
- Barabasi, A.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Maynard Smith, J. Natural selection and the concept of protein space. Nature 1970, 225, 563–564. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P.; Fontana, W.; Stadler, P.; Hofacker, I. From sequences to shapes and back: A case-study in RNA secondary structures. Proc. R. Soc. Lond. B 1994, 255, 279–284. [Google Scholar] [CrossRef]
- Bornberg-Bauer, E.; Chan, H.S. Modeling evolutionary landscapes: Mutational stability, topology and superfunnels in sequence space. Proc. Natl. Acad. Sci. USA 1999, 96, 10689–10694. [Google Scholar] [CrossRef] [PubMed]
- Van Nimwegen, E.; Crutchfield, J.P.; Huynen, M. Neutral evolution of mutational robustness. Proc. Natl. Acad. Sci. USA 1999, 96, 9716–9720. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.O. Adaptive evolution on neutral networks. Bull. Math. Biol. 2001, 63, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Bastolla, U.; Farwer, J.; Knapp, E.W.; Vendruscolo, M. How to guarantee optimal stability for most representative structures in the protein data bank. Proteins 2001, 44, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.; Gerhart, J. Evolvability. Proc. Natl. Acad. Sci. USA 1998, 95, 8420–8427. [Google Scholar] [CrossRef] [PubMed]
- Freeland, S.J. The Darwinian genetic code: an adaptation for adapting? Genet. Program. Evol. Mach. 2002, 3, 113–127. [Google Scholar] [CrossRef]
- Zhu, W.; Freeland, S. The standard genetic code enhances adaptive evolution of proteins. J. Theor. Biol. 2006, 239, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A. Robustness and Evolvability in Living Systems; Princeton University Press: Princeton, NJ, USA, 2007. [Google Scholar]
- Masel, J.; Trotter, M.V. Robustness and evolvability. Trends Genet. 2010, 9, 406–414. [Google Scholar] [CrossRef]
- Viney, M.; Reece, S.E. Adaptive noise. Proc. R. Soc. Lond. B 2013, 280. [Google Scholar] [CrossRef]
- Sueoka, N. On the genetic basis of variation and heterogeneity of DNA base composition. Proc. Natl. Acad. Sci. USA 1962, 48, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Lang, B.F. Parallels in genome evolution in mitochondria and bacterial symbionts. IUBMB Life 2003, 55, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. The Proteomic Constraint and its role in molecular evolution. Mol. Biol. Evol. 2008, 25, 2557–2565. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, A.; Rivera-Rivera, R.; Massey, S.E. The presence of the DNA repair genes mutM, mutY, mutL and mutS is related to proteome size in bacterial genomes. Front. Evol. Genomic Microbiol. 2012, 3. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, A.; Alicea, M.; Vicens, L.; Massey, S.E. The distribution of recombination repair genes is linked to information content in bacteria. Gene 2013, 528, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Mendez, R.; Fritsche, M.; Porto, M.; Bastolla, U. Mutation bias favors protein folding stability in the evolution of small populations. PLoS Comput. Biol. 2010, 6, e1000767. [Google Scholar] [CrossRef] [PubMed]
- Light, S.; Kraulis, P.; Elofsson, A. Preferential attachment in the evolution of metabolic networks. BMC Genomics 2005, 6. [Google Scholar] [CrossRef]
- Edwards, J.S.; Palsson, B.O. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J. Biol. Chem. 1999, 274, 17410–17416. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Palsson, B.O. Robustness analysis of the Escherichia coli metabolic network. Biotech. Prog. 2000, 16, 927–939. [Google Scholar] [CrossRef]
- Wagner, A. How the global structure of protein interaction networks evolves. Proc. R. Soc. Lond. B 2003, 270, 457–466. [Google Scholar] [CrossRef]
- Berg, J.; Lassig, M.; Wagner, A. Structure and evolution of protein interaction networks: A statistical model for link dynamics and gene duplications. BMC Evol. Biol. 2004, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, J.; Ouyang, S.; Wang, J.; Wu, S.; Wan, P.; Zhu, Y.; Xu, X.; He, F. Protein interaction networks of Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster: Large-scale organization and robustness. Proteomics 2006, 6, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W.; Conant, G.C.; Wagner, A. Molecular evolution in large genetic networks: Does connectivity equal constraint. J. Mol. Evol. 2004, 58, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, S.A.; Badu, M.M. Gene network regulatory growth by duplication. Nat. Genet. 2004, 36, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.D.J.; van Mourik, S.; van Ham, R.C.H.J. Mutational robustness of gene regulatory networks. PLoS One 2012, 7, e30591. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.O.; Wang, J.L.; Ofria, C.; Lenski, R.E.; Adami, C. Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature 2001, 412, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Szollosi, G.J.; Derenyi, I. The effect of recombination on the neutral evolution of genetic robustness. Math. Biosci. 2008, 214, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Taverna, D.M.; Goldstein, R.A. Why are proteins so robust to site mutations? J. Mol. Biol. 2002, 315, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Physiological and evolutionary theories of dominance. Am. Nat. 1934, 68, 25–53. [Google Scholar]
- Kacser, H.; Burns, J.A. The molecular basis of dominance. Genetics 1981, 97, 639–666. [Google Scholar] [PubMed]
- Mattimore, V.; Battista, J.R. Radioresistance of Deinococcus radiodurans: Functions necessary to survive ionizing radiation are also necessary to survive prolonged dessication. J. Bacteriol. 1996, 178, 633–637. [Google Scholar] [PubMed]
- Bernstein, H.; Byers, G.S.; Michod, R.E. Evolution of sexual reproduction: Importance of DNA repair, complementation, and variation. Am. Nat. 1981, 117, 537–549. [Google Scholar] [CrossRef]
- Belshaw, R.; Gardner, A.; Rambaut, A.; Pybus, O.G. Pacing a small cage: Mutation and RNA viruses. Trends Ecol. Evol. 2008, 23, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Silander, O.K.; Weinreich, D.M.; Wright, K.M.; O’Keefe, K.J.; Rang, C.U.; Turner, P.E.; Chao, L. Widespread genetic exchange among terrestrial bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 19009–19014. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Deutsch, M.; Wang, W.; Betran, E.; Brunet, F.G.; Zhang, J. Origin of new genes: Evidence from experimental and computational analysis. Genetica 2003, 118, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Chorev, M.; Carmel, L. Computational identification of functional introns: High positional conservation of introns that harbor RNA genes. Nucleic Acids Res. 2013, 41, 5604–5613. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Bashton, M.; Kerrison, N.D.; Chothia, C.; Teichmann, S.A. Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 2004, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Rozen, D.E.; Habets, M.G.J.L.; Handel, A.; de Visser, J.A.G.M. Heterogenous adaptive trajectories of small populations on complex fitness landscapes. PLoS One 2007, 3, e1715. [Google Scholar] [CrossRef]
- Jain, K.; Krug, J.; Park, S-C. Evolutionary advantage of small populations on complex fitness landscapes. Evolution 2011, 65, 1945–1955. [Google Scholar]
- Graur, D.; Zhang, Y.; Price, N.; Azevedo, R.B.R.; Zufall, R.A.; Elhaik, E. On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol. Evol. 2013, 5, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M.; Kreitman, M. The correlation between intron length and recombination in Drosophila: Dynamic equilibrium between mutational and selective forces. Genetics 2000, 156, 1175–1190. [Google Scholar] [PubMed]
- Roze, D.; Barton, N. The Hill-Robertson effect and the evolution of recombination. Genetics 2006, 173, 1793–1811. [Google Scholar] [CrossRef] [PubMed]
- Comeron, J.M.; Williford, A.; Kliman, R.M. The Hill–Robertson effect: Evolutionary consequences of weak selection and linkage in finite populations. Heredity 2008, 100, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Quietsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.; Siegal, M.L. Evolutionary capacitance as a general feature of complex gene networks. Nature 2003, 424, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueda, T.; Watanabe, K. The “polysemous” codon—A codon with multiple amino acid assignment caused by dual specificity of tRNA identity. EMBO J. 1997, 16, 899–1134. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.M.; Paredes, J.A.; Moura, G.R.; Manadas, B.; Lima-Costa, T.; Rocha, R.; Miranda, I.; Gomes, A.C.; Koerkamp, M.J.G.; Perrot, M.; et al. Critical roles for a genetic code alteration in the evolution of the genus Candida. EMBO J. 2007, 26, 4555–4565. [Google Scholar]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Ann. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.A.; van de Peer, Y. Angiosperm polyploids and their road to evolutionary success. Trends Evol. Biol. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Michod, R.E.; Wojciechowski, M.F.; Hoelzer, M.A. DNA repair and the evolution of transformation in the bacterium Bacillus subtilis. Genetics 1988, 118, 31–39. [Google Scholar] [PubMed]
- Finkel, S.E.; Kolter, R. DNA as a nutrient: Novel role for bacterial competence gene homologs. J. Bacteriol. 2001, 183, 6288–6293. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.; Sundstrom, A.; Massey, S.E.; Mishra, B. What can information-asymmetric games tell us about the context of Crick’s “frozen accident”? J. R. Soc. Interface 2013, 10, 20130614. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.; Diamond, A. UGA: A split personality in the universal genetic code. Trends Genet. 1993, 9, 69–70. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, P.O.; Prat, L.; Heinemann, I.U.; Ling, J.; Odoi, K.; Liu, W.R.; Soll, D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNAPyl for genetic code expansion. FEBS Lett. 2012, 586, 3931–3937. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, D. Tryptophan transfer RNA as the UGA suppressor. Trends Genet. 1970, 58, 439–444. [Google Scholar]
- Moran, N. Microbial minimalism: genome reduction in bacterial pathogens. Cell 2002, 108, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Andachi, Y.; Yamao, F.; Muto, A.; Osawa, S. Codon recognition patterns as deduced from sequences of the complete set of transfer RNA species in Mycoplasma capricolum. Resemblance to mitochondria. J. Mol. Biol. 1989, 209, 37–54. [Google Scholar]
- Weisburg, W.G.; Tully, J.G.; Rose, D.L.; Petzel, J.P.; Oyaizu, H.; Yang, D.; Mandelco, L.; Sechrest, J.; Lawrence, T.G.; Van Etten, J. A phylogenetic analysis of the mycoplasmas: Basis for their classification. J. Bacteriol. 1989, 171, 6455–6467. [Google Scholar] [PubMed]
- Carvalho, F.M.; Fonseca, M.M.; de Medeiros, S.B.; Scortecci, K.C.; Blaha, C.A.; Agnez-Lima, L.F. DNA repair in reduced genome: The Mycoplasma model. Gene 2005, 360, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Citti, C.; Marechal-Drouard, L.; Saillard, C.; Weil, J.H.; Bove, J.M. Spiroplasma citri UGG and UGA tryptophan codons: Sequence of the two tryptophanyl-tRNAs and organization of the corresponding genes. J. Bacteriol. 1992, 174, 6471–6478. [Google Scholar] [PubMed]
- Carle, P.; Laigret, F.; Tully, J.G.; Bove, J.M. Heterogeneity of genome sizes within the genus Spiroplasma. Int. J. Syst. Bacteriol. 1995, 45, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.; Lo, W.-S.; Chen, L.-L.; Kuo, C.-H. Complete genomes of two dipteran-associated Spiroplasmas provided insights into the origin, dynamics and impacts of viral invasion in Spiroplasma. Genome Biol. Evol. 2013, 5, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.-S.; Chen, L.-L.; Chung, W.-C.; Gasparich, G.E.; Kuo, C.-H. Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, A. Ureaplasma urealyticum urease genes; use of a UGA tryptophan codon. Mol. Microbiol. 1990, 4, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Paralanov, V.; Lu, J.; Duffy, L.; Crabb, D.; Shrivastava, S.; Methe, B.; Inman, J.; Yooseph, S.; Xiao, L.; Cassell, G.; et al. Comparative genome analysis of 19 Ureaplasma urealyticum and Ureaplasma parvum strains. BMC Microbiol. 2012, 12. [Google Scholar] [CrossRef]
- Campbell, J.H.; O’Donoghue, P.; Campbell, A.G.; Schwientek, P.; Sczyrba, A.; Woyke, T.; Soll, D.; Podar, M. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA 2013, 110, 5540–5545. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.S.; Wrighton, K.C.; Handley, K.M.; Sharon, I.; Hug, L.A.; Castelle, C.J.; Thomas, B.C.; Banfield, J.F. Small Genomes and Sparse Metabolisms of Sediment-Associated Bacteria from Four Candidate Phyla. mBio 2013, 4. [Google Scholar] [CrossRef]
- Bennett, G.M.; Moran, N.A. Small, smaller, smallest: The origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 2013, 5, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19392–19397. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; McDonald, B.R.; Moran, N.A. Origin of an alternative genetic code in the extremely small and GC-rich genome of a bacterial symbiont. PLoS Genet. 2009, 5, e1000565. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ishikawa, S.A.; Hashimoto, T.; Inagaki, Y. A deviant genetic code in the green alga-derived plastid in the dinoflagellate Lepidodinium chlorophorum. Mol. Phylogent. Evol. 2011, 60, 68–72. [Google Scholar] [CrossRef]
- Ohama, T.; Suzuki, T.; Mori, M.; Osawa, S.; Ueda, T.; Watanabe, K.; Nakase, T. Non-universal decoding of the leucine codon CUG in several Candida species. Nucleic Acids Res 1993, 21, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.S.; Tuite, M.F. The CUG codon is decoded in vivo as serine and not leucine in Candida albicans. Nucleic Acids Res. 1995, 23, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Doolittle, W.F. A non-canonical genetic code in an early diverging eukaryotic lineage. EMBO J. 1996, 15, 2285–2290. [Google Scholar] [PubMed]
- Keeling, P.J.; Doolittle, W.F. Widespread and ancient distribution of a noncanonical genetic code in diplomonads. Mol. Biol. Evol. 1997, 14, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Leander, B.S. Characterization of a non-canonical genetic code in the oxymonad Streblomastix strix. J. Mol. Biol. 2006, 326, 1337–1349. [Google Scholar] [CrossRef]
- Kuchino, Y.; Hanyu, N.; Tashiro, F.; Nishimura, S. Tetrahymena thermophila glutamine tRNA and its gene that corresponds to UAA termination codon. Proc. Natl. Acad. Sci. USA 1985, 82, 4758–4762. [Google Scholar] [CrossRef] [PubMed]
- Preer, J.R., Jr.; Preer, L.B.; Rudman, B.M.; Barnett, A.J. Deviations from the universal code shown by the gene for surface protein 51A in Paramecium. Nature 1985, 314, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Schmidt, H.J.; Plumper, E.; Hasilik, A.; Mersmann, G.; Meyer, H.E.; Engstrom, A.; Heckmann, K. UGA is translated as cysteine in pheromone 3 of Euplotes octocarinatus. Proc. Natl. Acad. Sci. USA 1991, 88, 3758–3761. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Knight, R.D.; Landweber, L.F. The molecular basis of nuclear genetic code change in ciliates. Curr. Biol. 2001, 11, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.U.; Leible, M.B.; Yang, X.P. Strong homology between the small subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase of two species of Acetabularia and the occurrence of unusual codon usage. Mol. Gen. Genet. 1989, 218, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.U.; de Groot, E.J. Sequences of two rbcS cDNA clones of Batophora oerstedii: Structural and evolutionary considerations. Curr. Genet. 1991, 20, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Gile, G.H.; Novis, P.; Cragg, D.; Zuccarello, G.C.; Keeling, P.J. The distribution of elongation factor-1alpha (EF-1α), elongation factor-like (EFL), and a noncanonical genetic code in the Ulvophyceae: Discrete genetic characters support a consistent phylogenetic framework. J. Eukaryot. Microbiol. 2009, 56, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Cocquyt, E.; Gile, G.H.; Leilaert, F.; Verbruggen, H.; Keeling, P.J.; de Clerck, O. Complex phylogenetic distribution of a non-canonical genetic code in green algae. BMC Evol. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. Augustus: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, 309–312. [Google Scholar] [CrossRef]
- Massey, S.E. Proteome size as the major factor determining mutation rates. Proc. Natl. Acad. Sci. USA 2013, 110, 858–859. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 1966, 8, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.; Ackerman, M.S.; Miller, S.F.; Doak, T.G.; Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 18488–18492. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.; Ackerman, M.S.; Miller, S.F.; Doak, T.G.; Lynch, M. Reply to Massey: Drift does influence mutation rate evolution. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Gabriel, W. Mutation load and survival of small populations. Evolution 1990, 44, 1725–1737. [Google Scholar] [CrossRef]
- Nga, P.T.; Parquet, M.d.C.; Lauber, C.; Parida, M.; Nabeshima, T.; Yu, F.; Thuy, N.T.; Inoue, S.; Ito, T.; Okamoto, K.; Ichinose, A.; Snijder, E.J.; et al. Discovery of the First Insect Nidovirus, a Missing Evolutionary Link in the Emergence of the Largest RNA Virus Genomes. PLoS Pathog. 2011, 7, e1002215. [Google Scholar]
- Stevens, K.; Weynberg, K.; Beltas, C.; Brown, S.; Brownlee, C.; Brown, C.; Brown, M.T.; Schroeder, D.C. A novel evolutionary strategy revealed in the phaeoviruses. PLoS One 2014, 9, e86040. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The Hypercycle: A Principle of Self-Organization; Springer: Berlin/Heidelberg, Germany, 1979. [Google Scholar]
- Eigen, M. Natural selection: a phase transition? Biophys. Chem. 2000, 85, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef] [PubMed]
- White, E.P.; Ernest, S.K.M.; Kerkhoff, A.J.; Enquist, B.J. Relationships between body size and abundance in ecology. Trends Ecol. Evol. 2007, 22, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Batut, B.; Knibbe, C.; Marais, G.; Daubin, V. Reductive genome evolution at both ends of the bacterial population size spectrum. Nat. Rev. Microbiol. 2014, 12, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, N.; Roggensack, S.E.; Rodrigue, S.; Thompson, J.W.; Biller, S.J.; Coe, A.; Ding, H.; Marttinen, P.; Malmstrom, R.R.; Stocker, R.; et al. Single-Cell Genomics Reveals Hundreds of Coexisting Subpopulations in Wild Prochlorococcus. Science 2014, 344, 416–420. [Google Scholar]
- Fisher, R.A. The Genetical Theory of Natural Selection; Oxford University Press: Oxford, UK, 1930. [Google Scholar]
- Springer, M.S.; Douzery, E. Secondary structure and patterns of evolution among mammalian mitochondrial 12S rRNA molecules. J. Mol. Evol. 1996, 43, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Comparative analysis of secondary structure of insect mitochondrial small subunit ribosomal RNA using maximum weighted matching. Nucleic Acids Res. 2000, 28, 3839–3845. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Suematsu, T.; Ohtsuki, T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Lynch, M. Mutation accumulation in transfer RNAs: Molecular evidence for Muller’s ratchet in mitochondrial genomes. Mol. Biol. Evol. 1996, 13, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol. Biol. Evol. 1997, 14, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Blanchard, J.L. Deleterious mutation accumulation in organelle genomes. Genetica 1998, 102–103, 29–39. [Google Scholar]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Felsenstein, J. The Evolutionary Advantage of Recombination. Genetics 1974, 78, 737–756. [Google Scholar] [PubMed]
- Haldane, J.B.S. On being the right size. In Possible Worlds and other Essays; Harper and Brothers: London, UK, 1928. [Google Scholar]
- Shannon, C.E. A mathematical theory of communication. Bell. System Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Ladyman, J.A.C.; Lambert, J.; Wiesner, K. What is a complex system? Eur. J. Phil. Sci. 2013, 3, 33–67. [Google Scholar] [CrossRef]
- Solomonoff, R. A formal theory of inductive inference Part I. Inf. Control 1964, 7, 1–22. [Google Scholar] [CrossRef]
- Solomonoff, R. A formal theory of inductive inference Part II. Inf. Control 1964, 7, 224–254. [Google Scholar] [CrossRef]
- Kolmogorov, A.N. Three approaches to quantitative definition of information. Probl. Inf. Transm. 1965, 1, 1–7. [Google Scholar]
- Jiang, Y.; Xu, C. The calculation of information and organismal complexity. Biol. Direct. 2010, 5. [Google Scholar] [CrossRef]
- D’Souza, N.; Holden, L.; Robson, S.; Mah, K.; Di Prospero, L.; Wong, C.S.; Chow, E.; Spayne, J. Modern palliative radiation treatment: Do complexity and workload contribute to medical errors? Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 43–48. [Google Scholar] [CrossRef]
- Taylor, J.M.G.; Siqueira, A.L.; Weiss, R.E. The cost of adding parameters to a model. J. R. Statist. Soc. B 1996, 58, 693–607. [Google Scholar]
- Fawcett, T.W.; Higgenson, A.D. Heavy use of equations impedes communication among biologists. Proc. Natl. Acad. Sci. USA 2012, 109, 11735–11739. [Google Scholar] [CrossRef] [PubMed]
- Promislow, D.E. DNA repair and the evolution of longevity: A critical analysis. J. Theor. Biol. 1994, 170, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Leroi, A.M.; Koufopanou, V.; Burt, A. Cancer selection. Nat. Rev. Cancer 2003, 3, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Caulin, A.F.; Maley, C.C. Peto’s paradox: Evolution’s prescription for cancer prevention. Trends Ecol. Evol. 2011, 26, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rivera, M.C.; Lake, J.A. Horizontal gene transfer among genomes: The complexity hypothesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.A. Adaptation and the cost of complexity. Evolution 2000, 54, 13–20. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massey, S.E. Genetic Code Evolution Reveals the Neutral Emergence of Mutational Robustness, and Information as an Evolutionary Constraint. Life 2015, 5, 1301-1332. https://doi.org/10.3390/life5021301
Massey SE. Genetic Code Evolution Reveals the Neutral Emergence of Mutational Robustness, and Information as an Evolutionary Constraint. Life. 2015; 5(2):1301-1332. https://doi.org/10.3390/life5021301
Chicago/Turabian StyleMassey, Steven E. 2015. "Genetic Code Evolution Reveals the Neutral Emergence of Mutational Robustness, and Information as an Evolutionary Constraint" Life 5, no. 2: 1301-1332. https://doi.org/10.3390/life5021301
APA StyleMassey, S. E. (2015). Genetic Code Evolution Reveals the Neutral Emergence of Mutational Robustness, and Information as an Evolutionary Constraint. Life, 5(2), 1301-1332. https://doi.org/10.3390/life5021301