
Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. What is Organocatalysis? The Organic Chemist’s View
3. Origin of Peptides
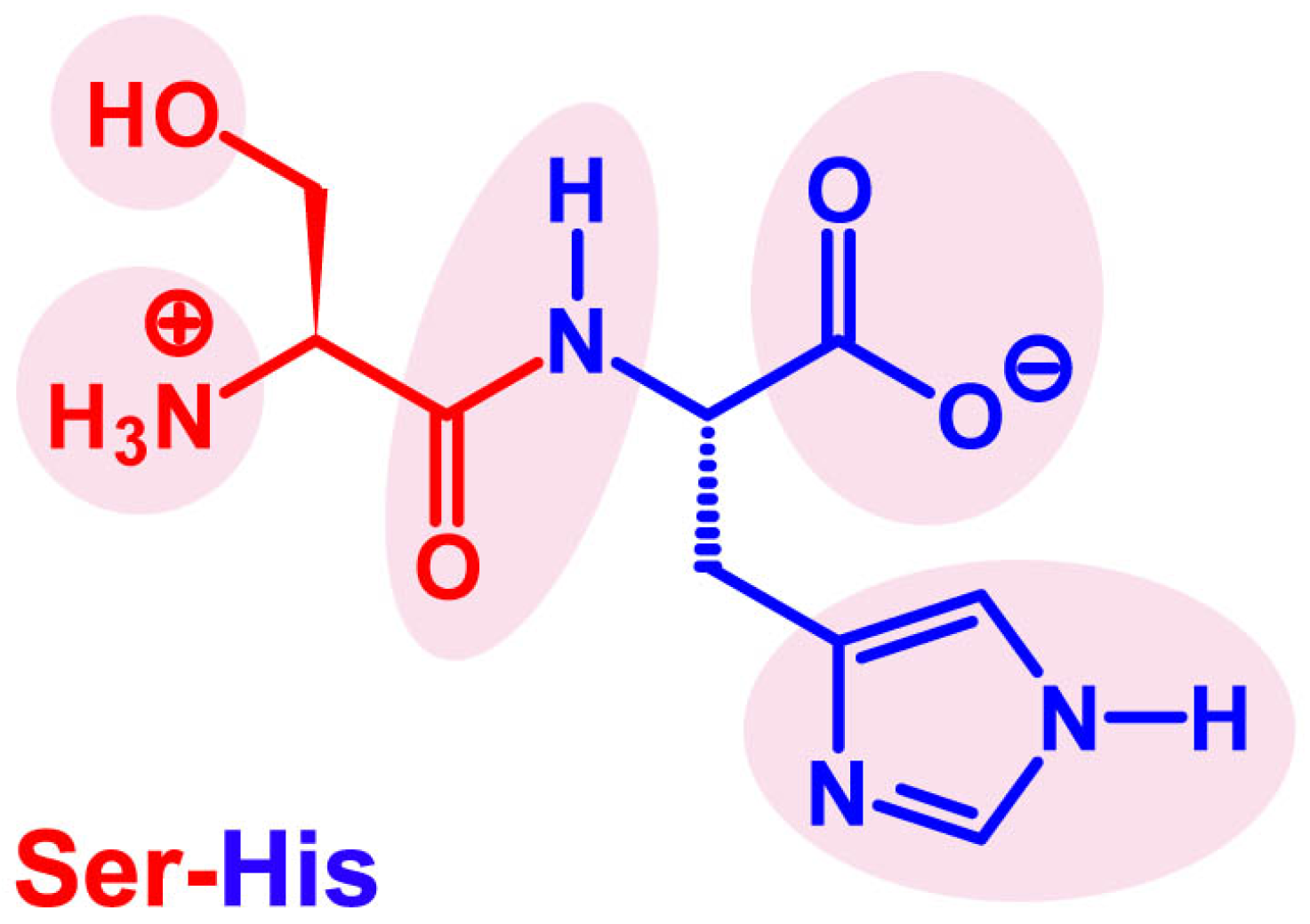
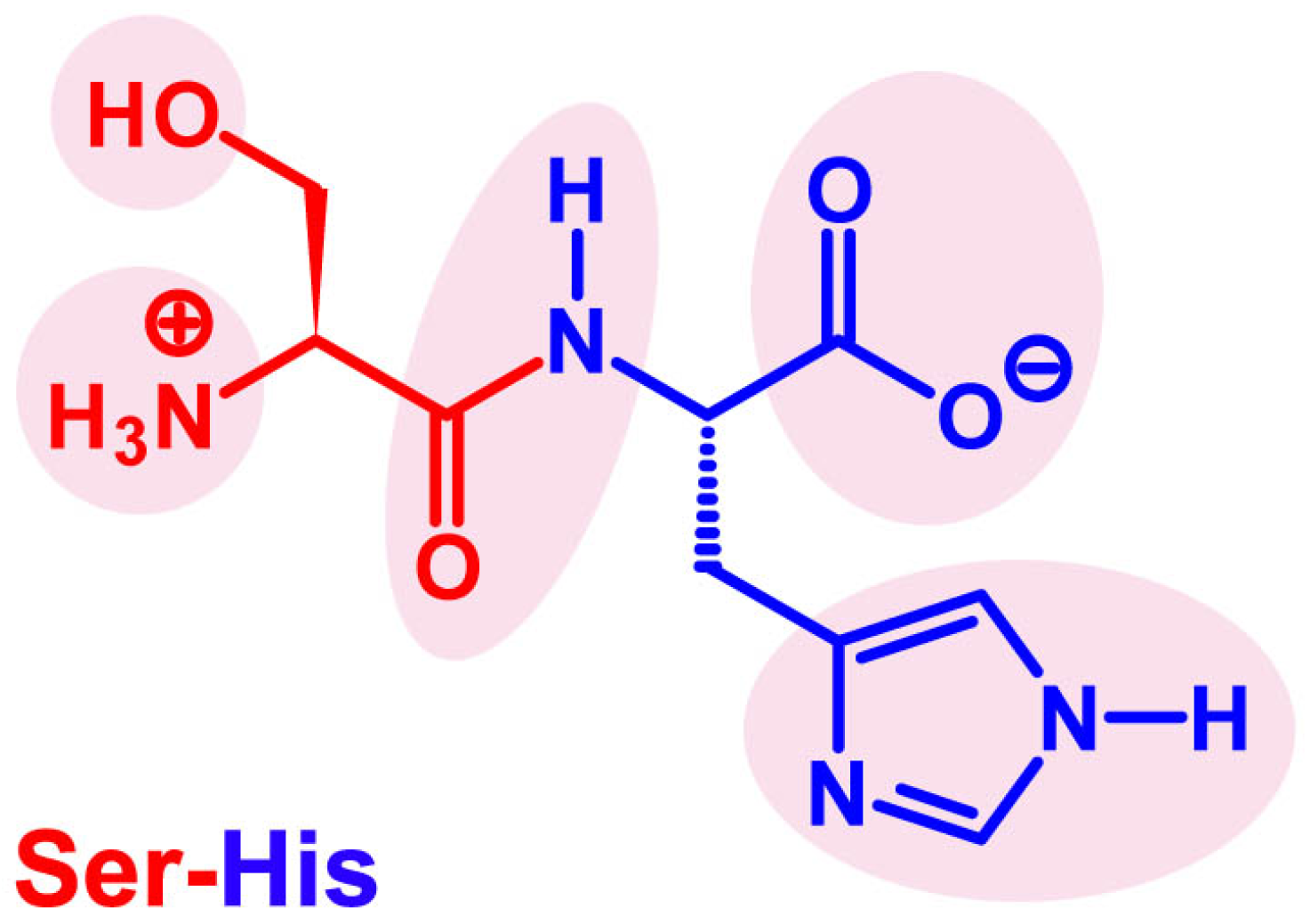
4. Seryl-Histidine Catalyzes the Formation of Peptide Bonds and Phosphodiester Bonds
4.1. The Discovery of Ser-His Hydrolase Activity
“In the studies of the interaction between N-phosphoamino acids with DNA, it was found that the aged solution of N-phosphorus serine in a saturated histidine buffer exhibited DNA cleavage activity, not the fresh one. Finally, it was clarified that the seryl-histidine dipeptide which formed in the solution was responsible for the DNA cleavage (Ma, 1996).”[80]
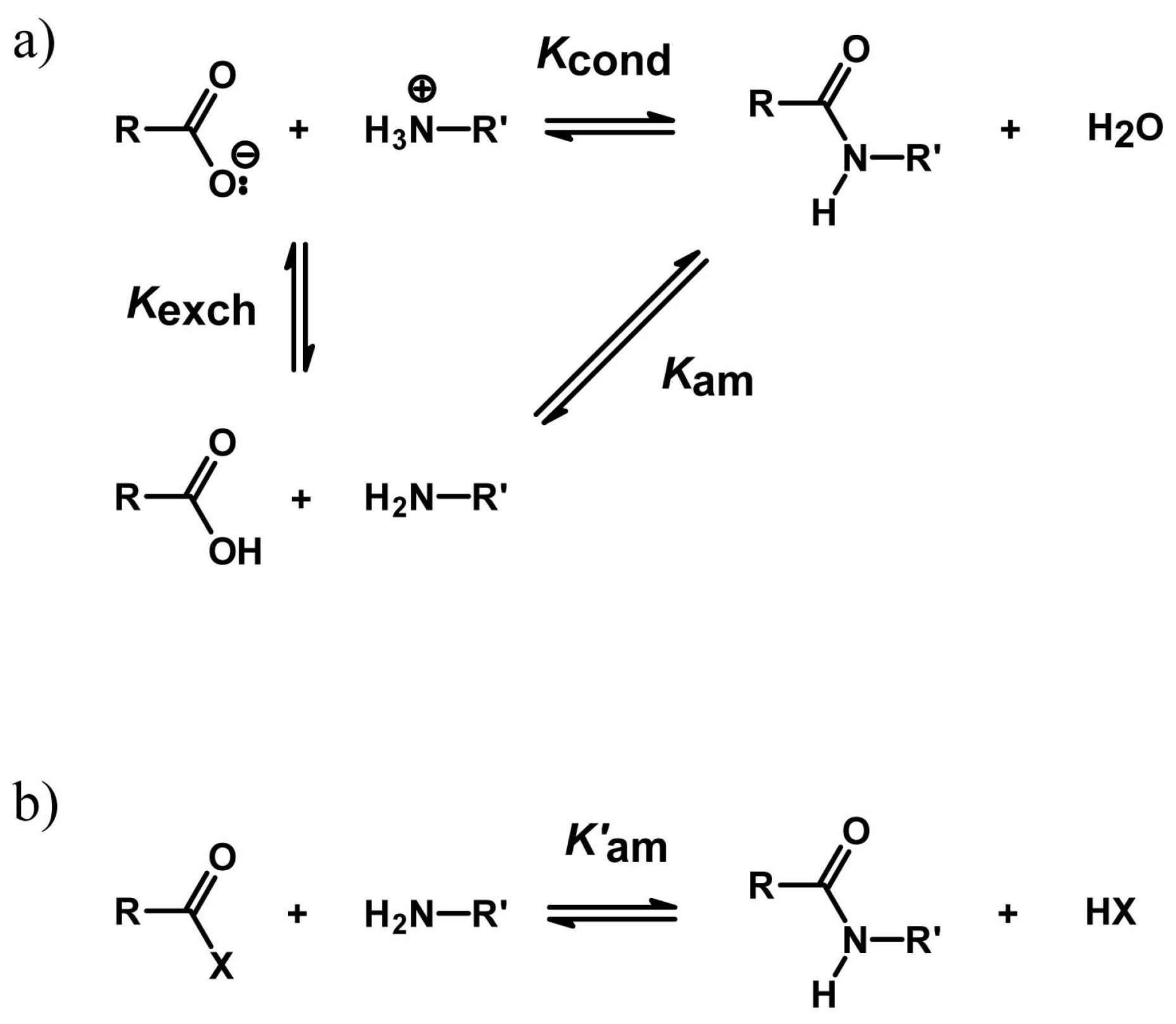
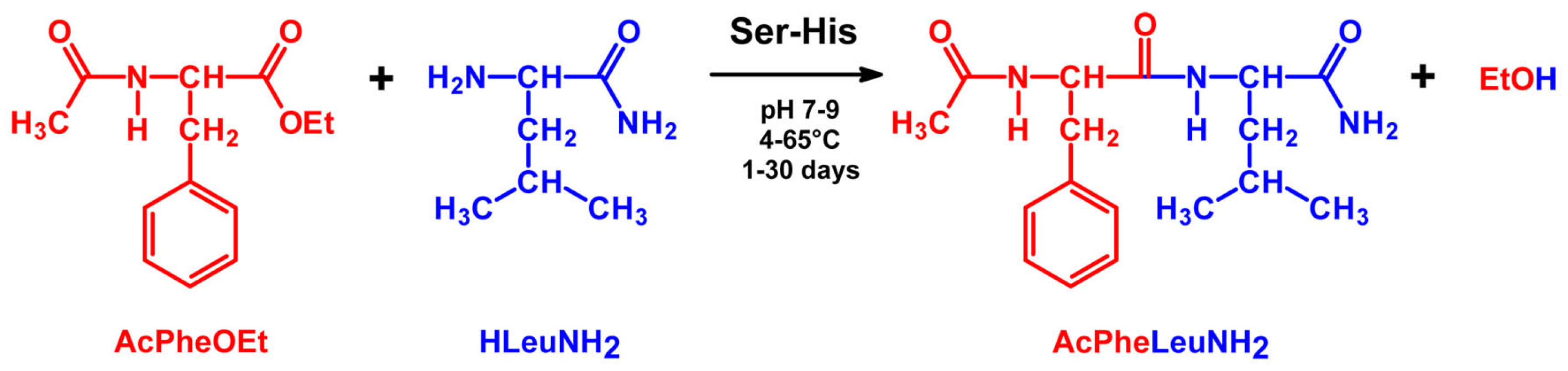
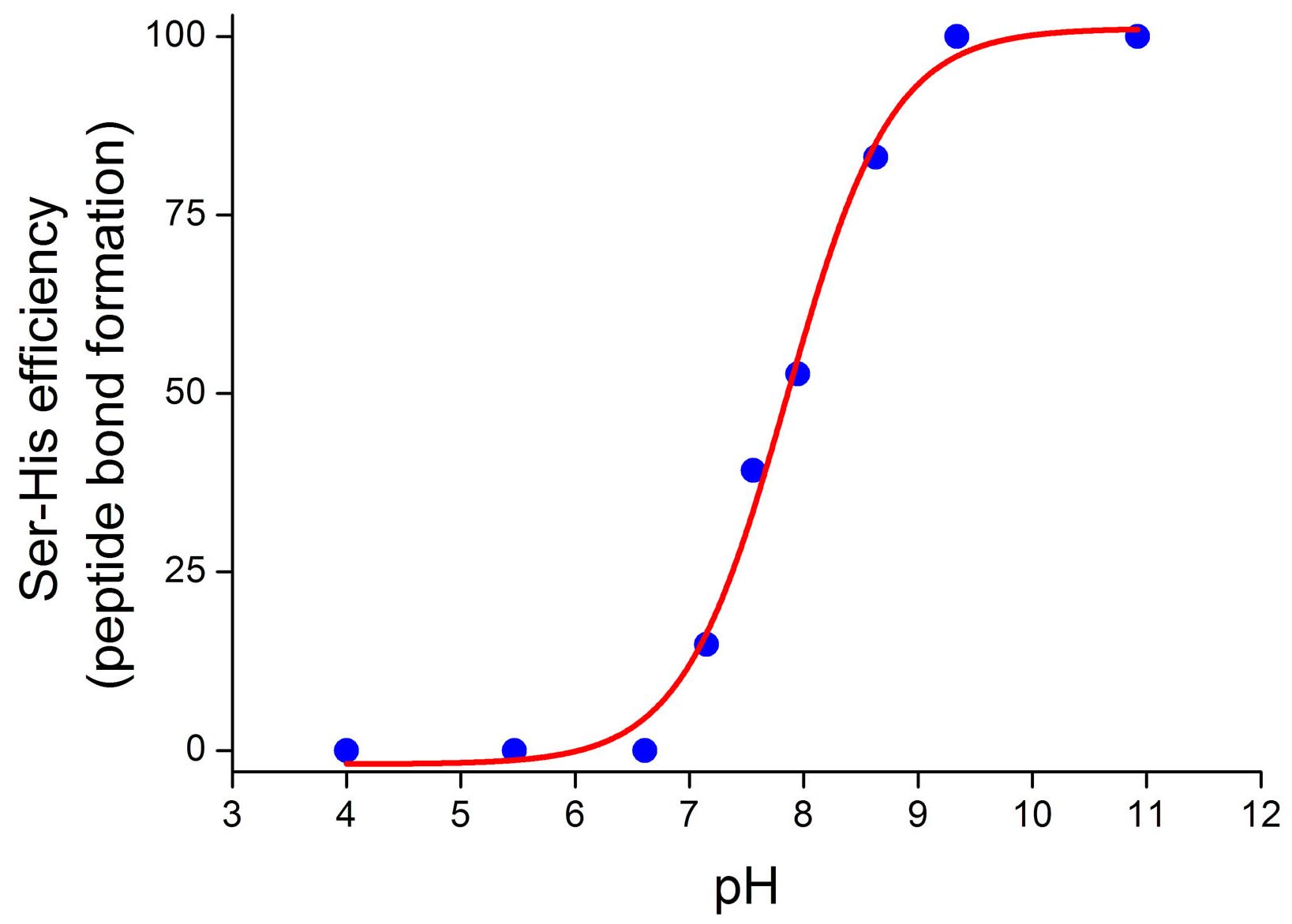
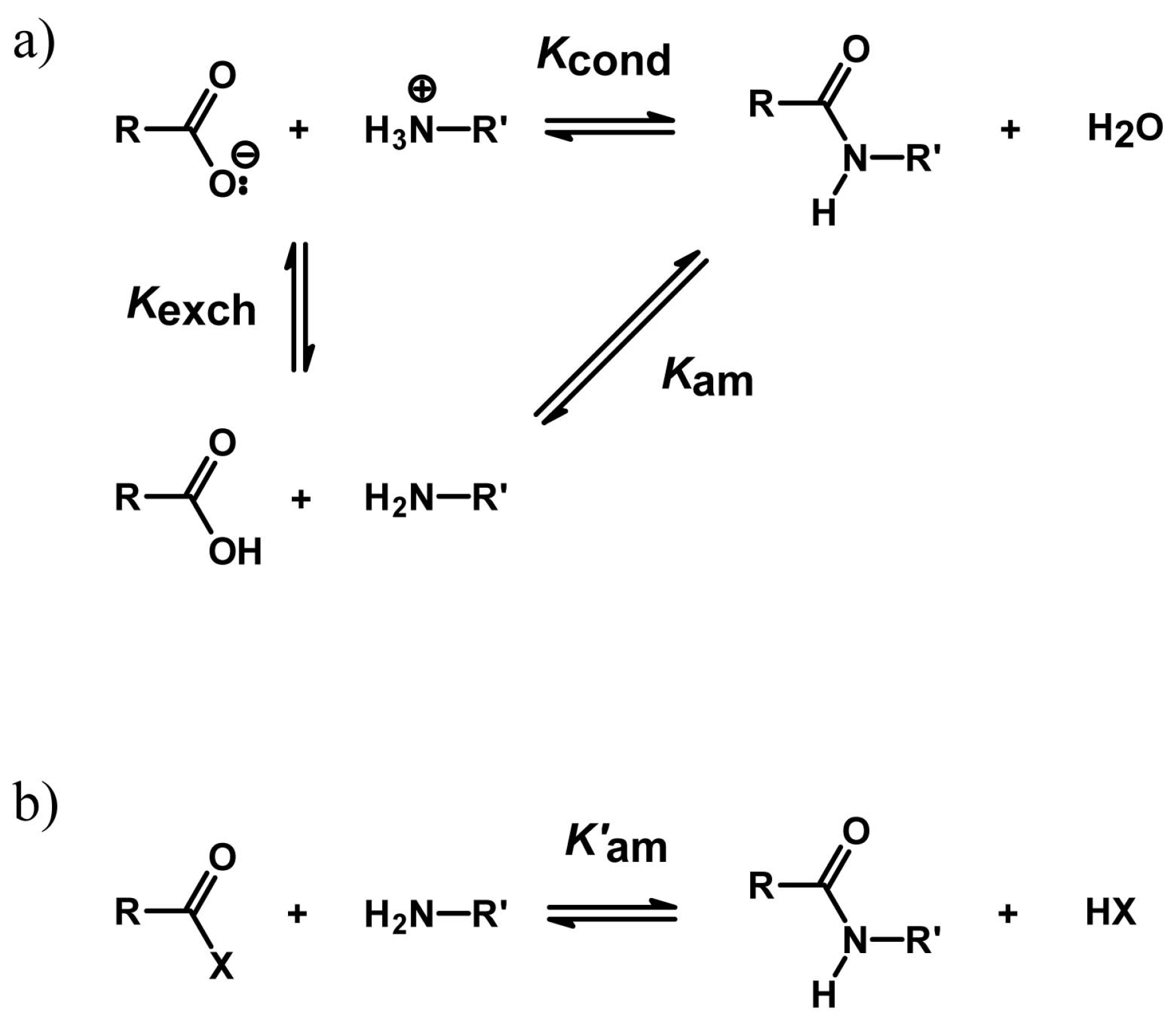
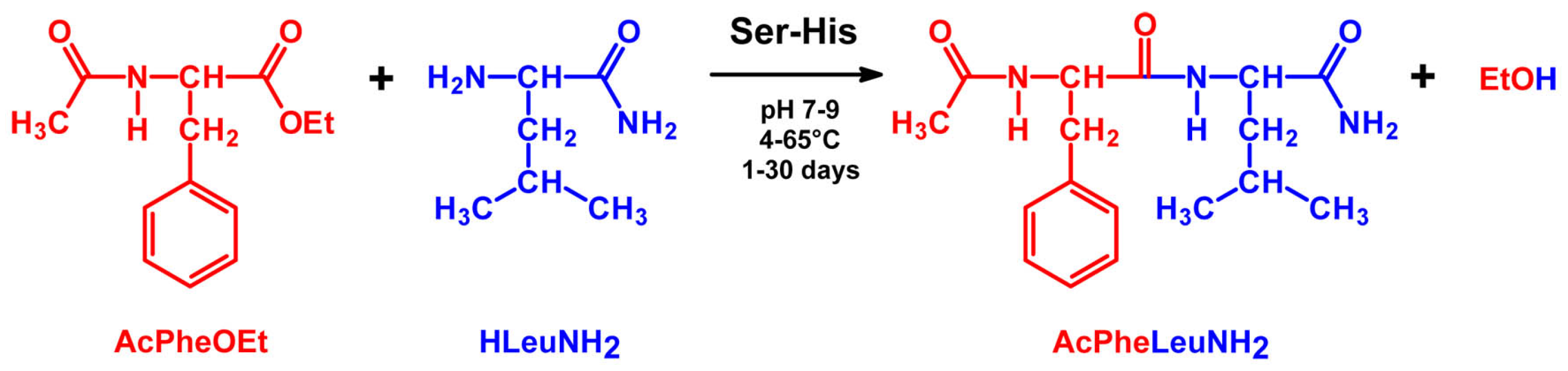
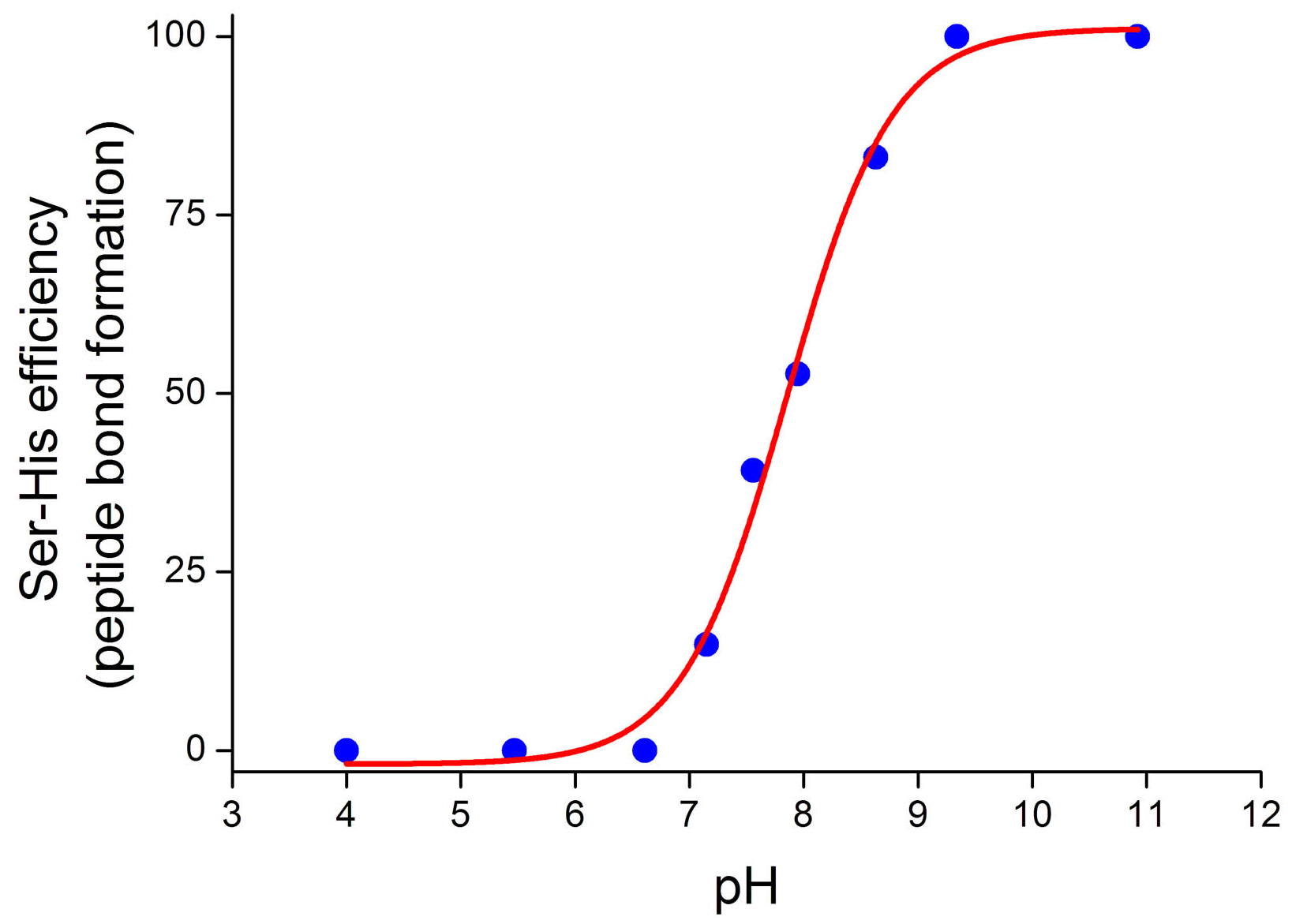
4.2. Peptide Bond Formation
4.3. Phosphodiester Bond Formation
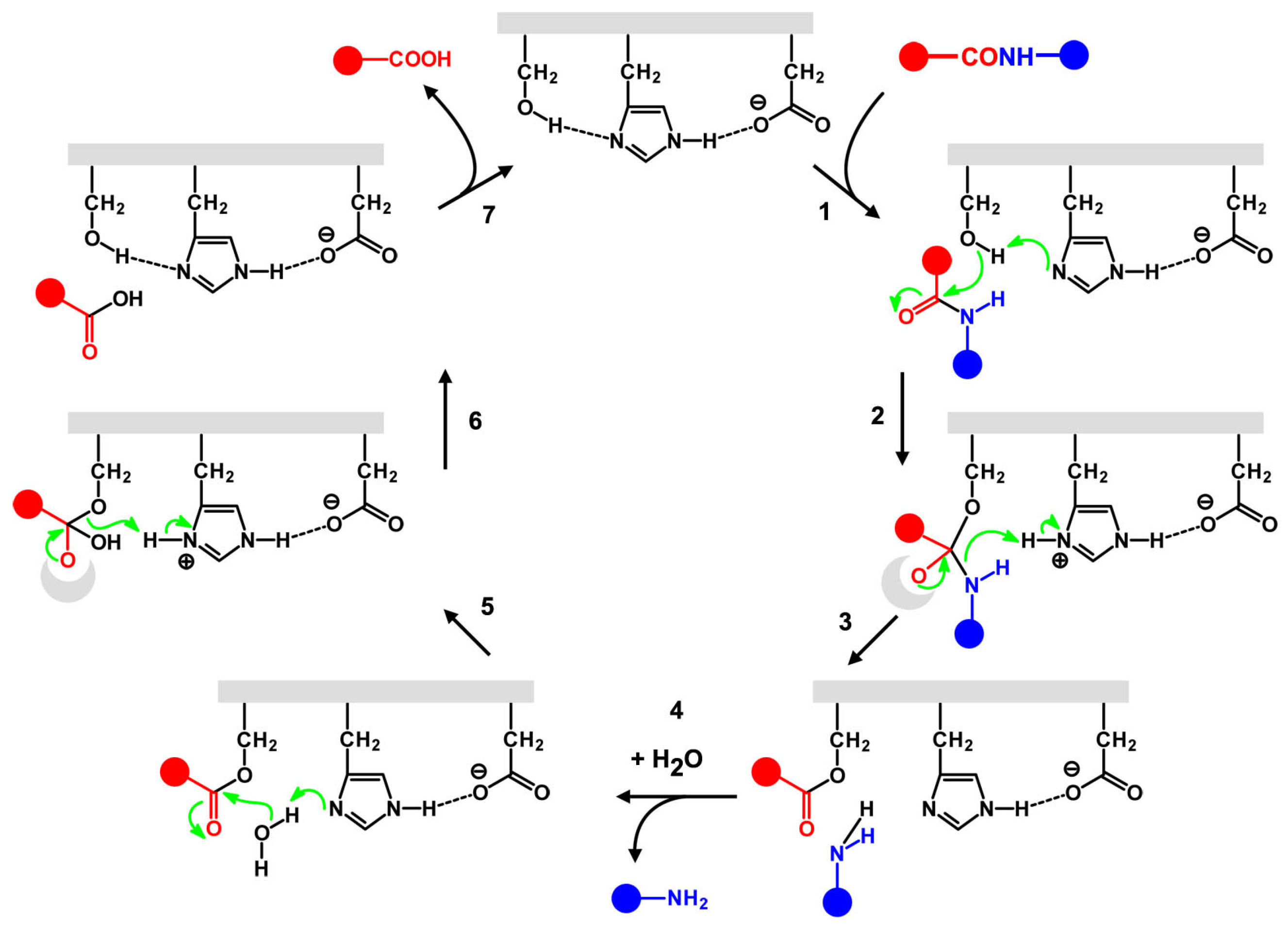
4.4. A Tentative Discussion on the Catalytic Mechanism of Ser-His
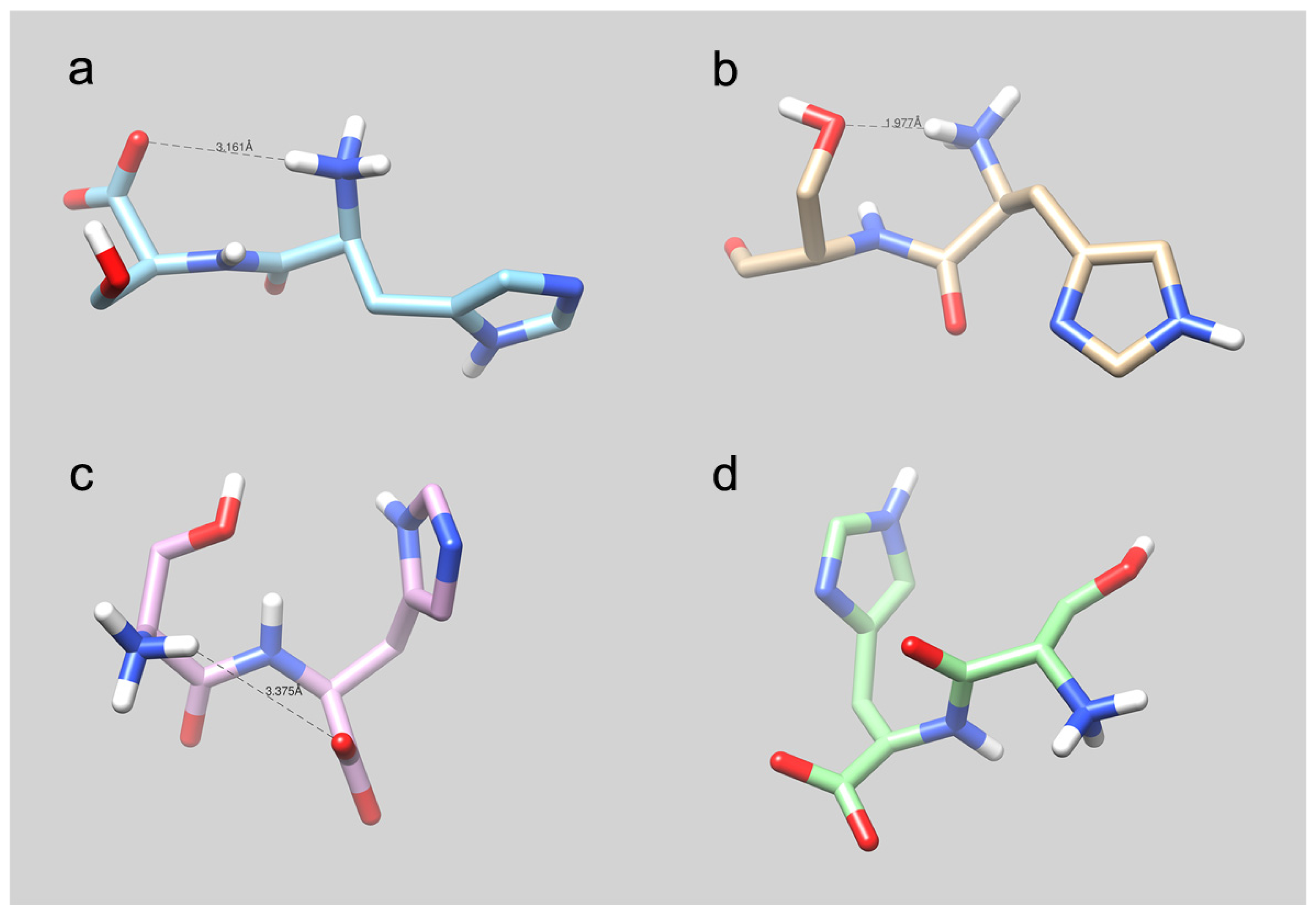
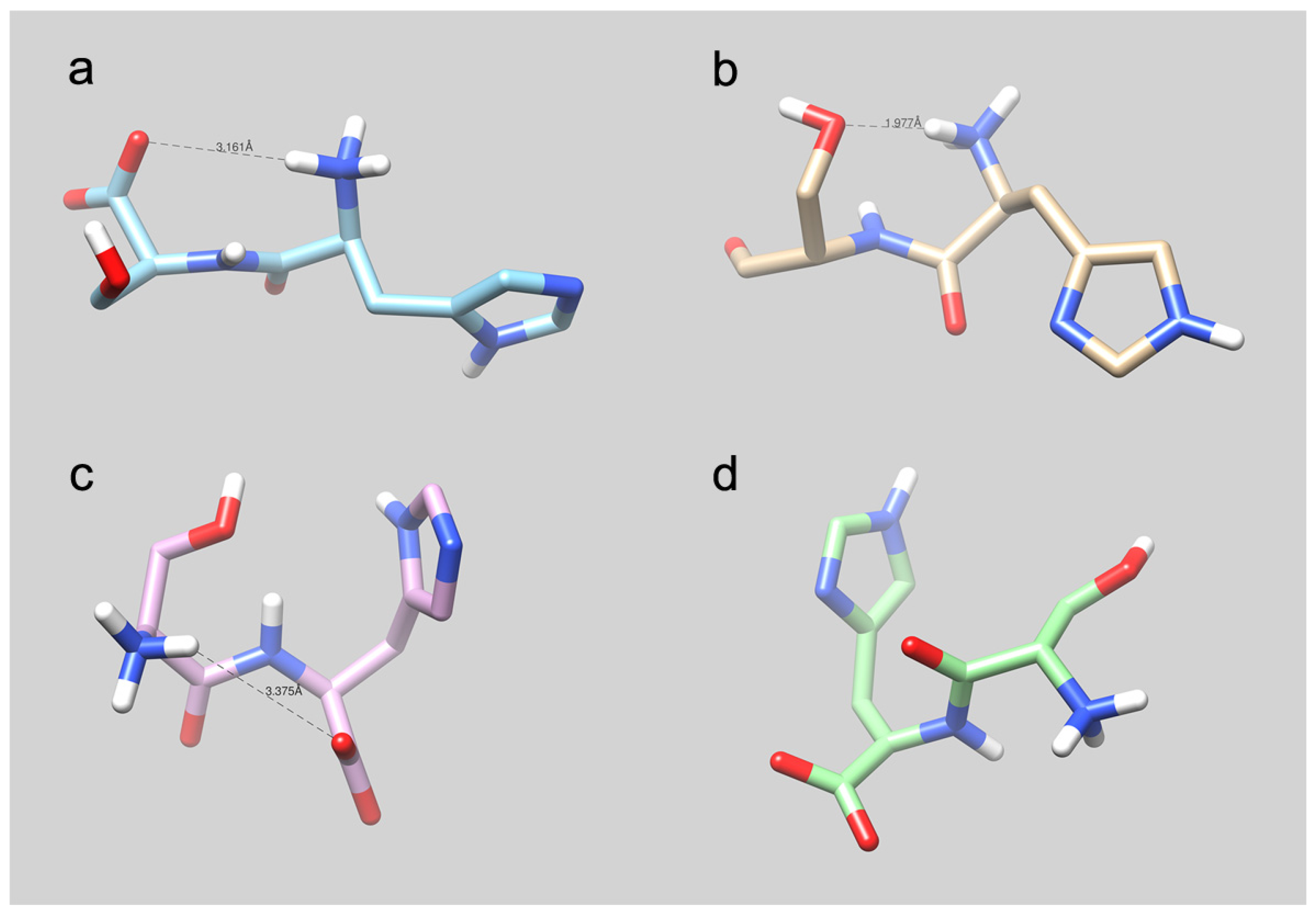
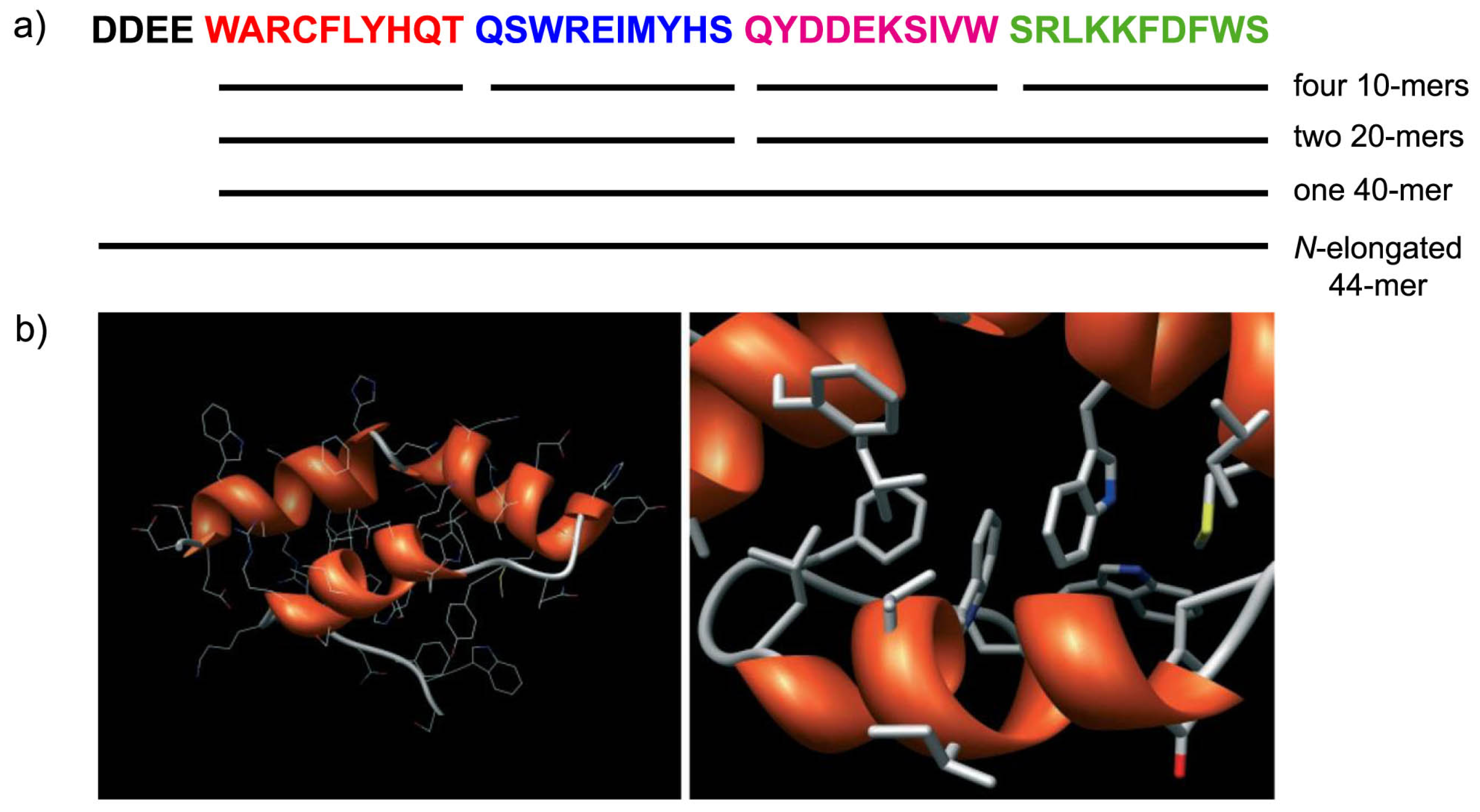
4.5. Molecular Modeling Studies: What Can We Learn?
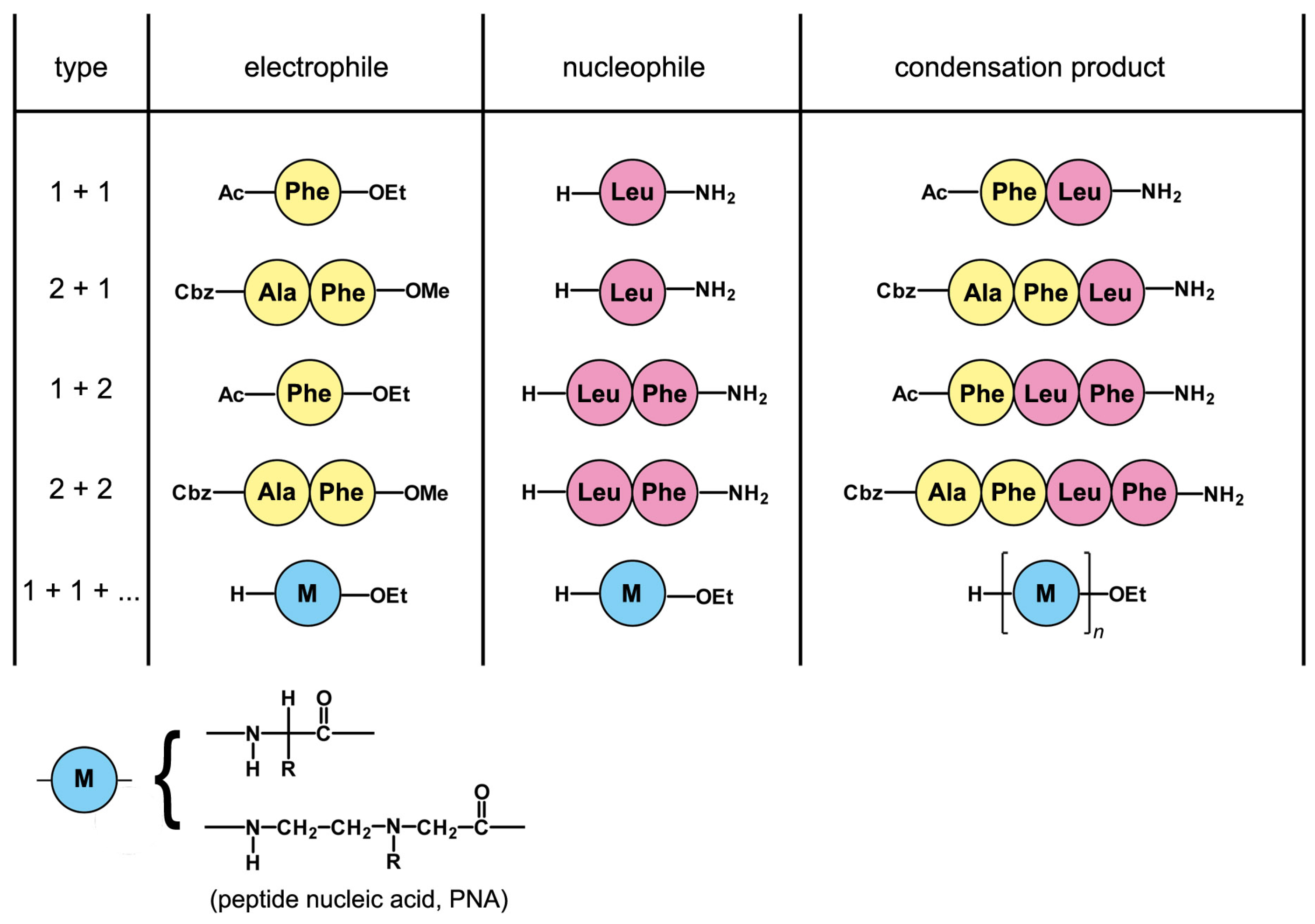
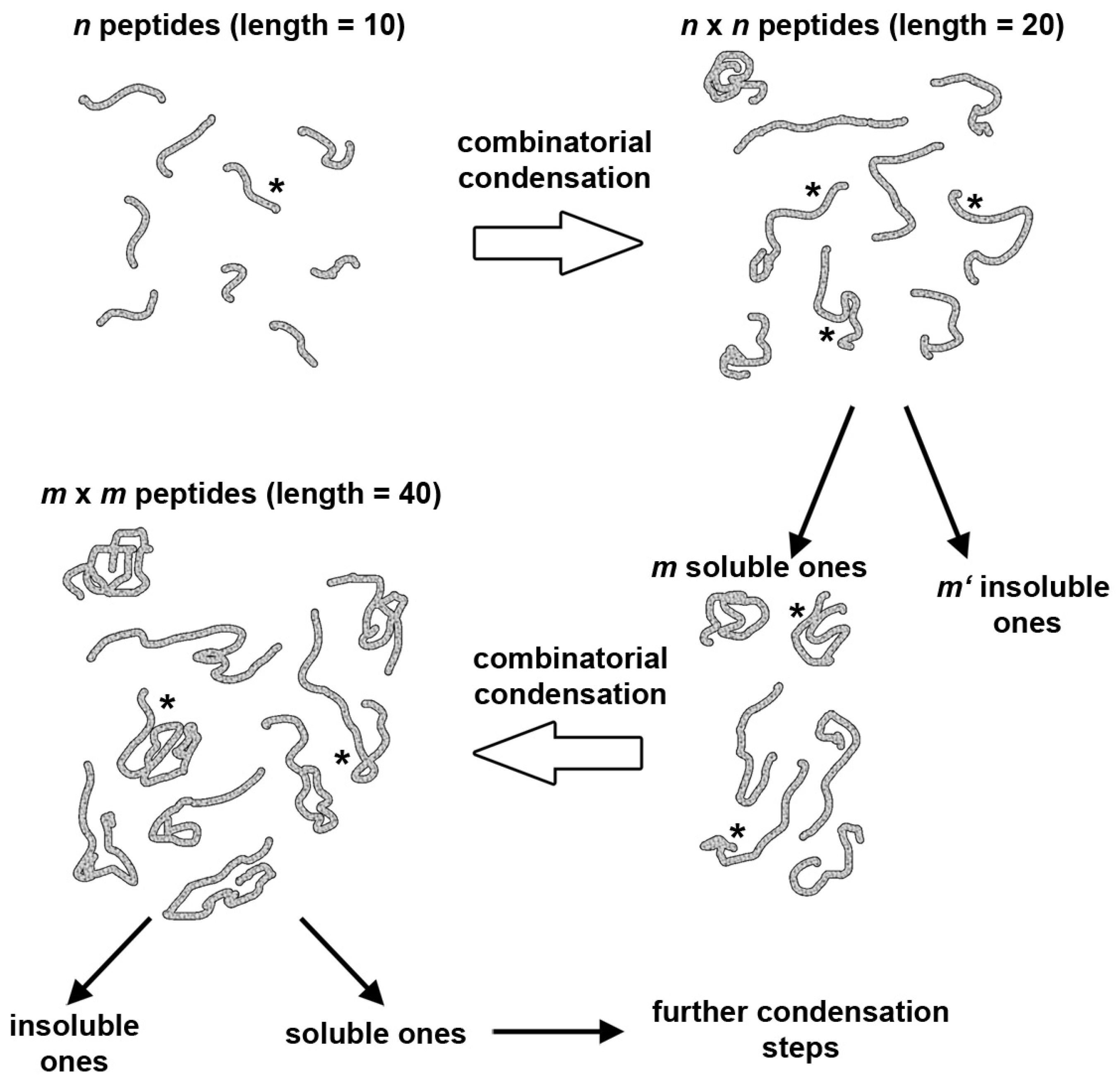
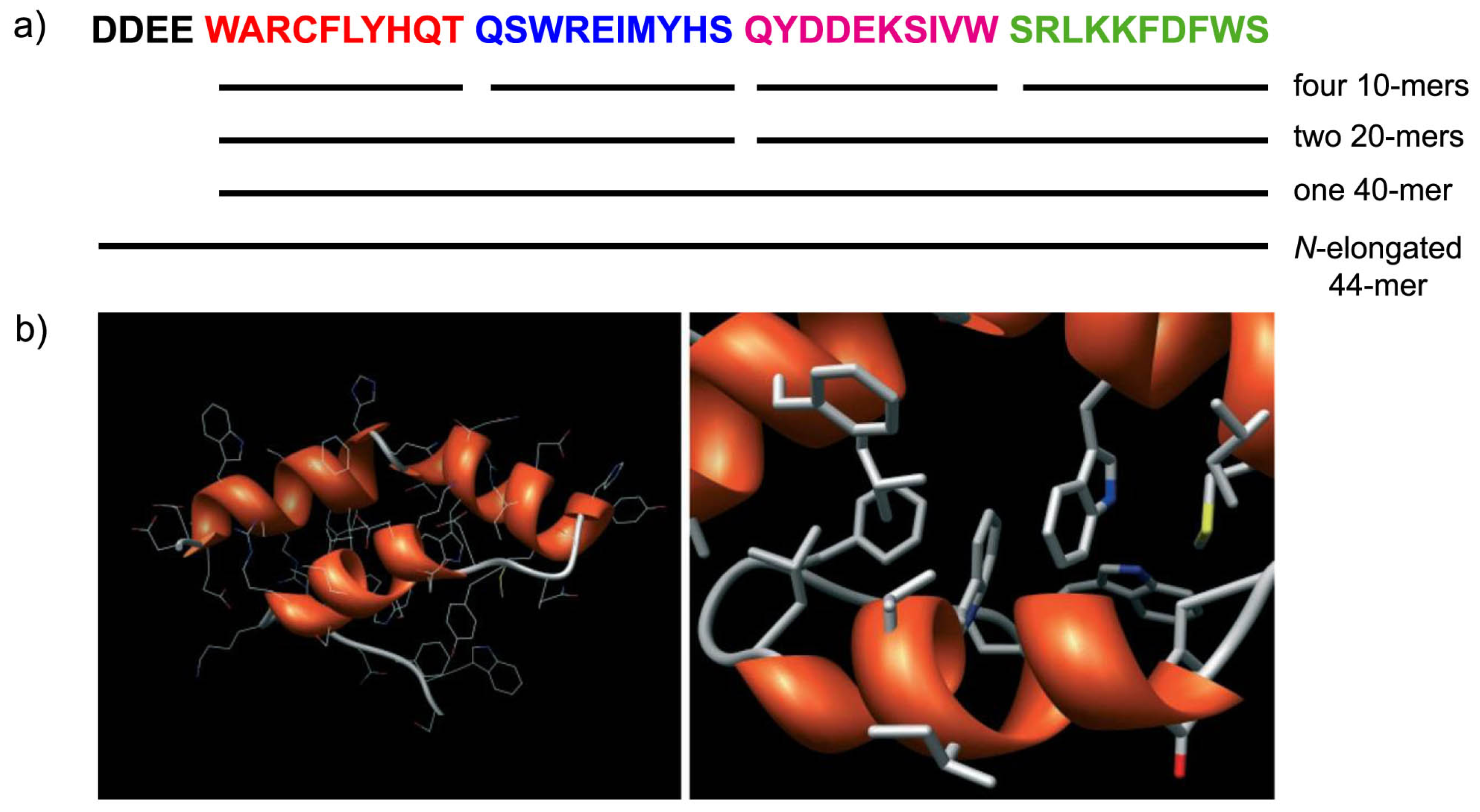
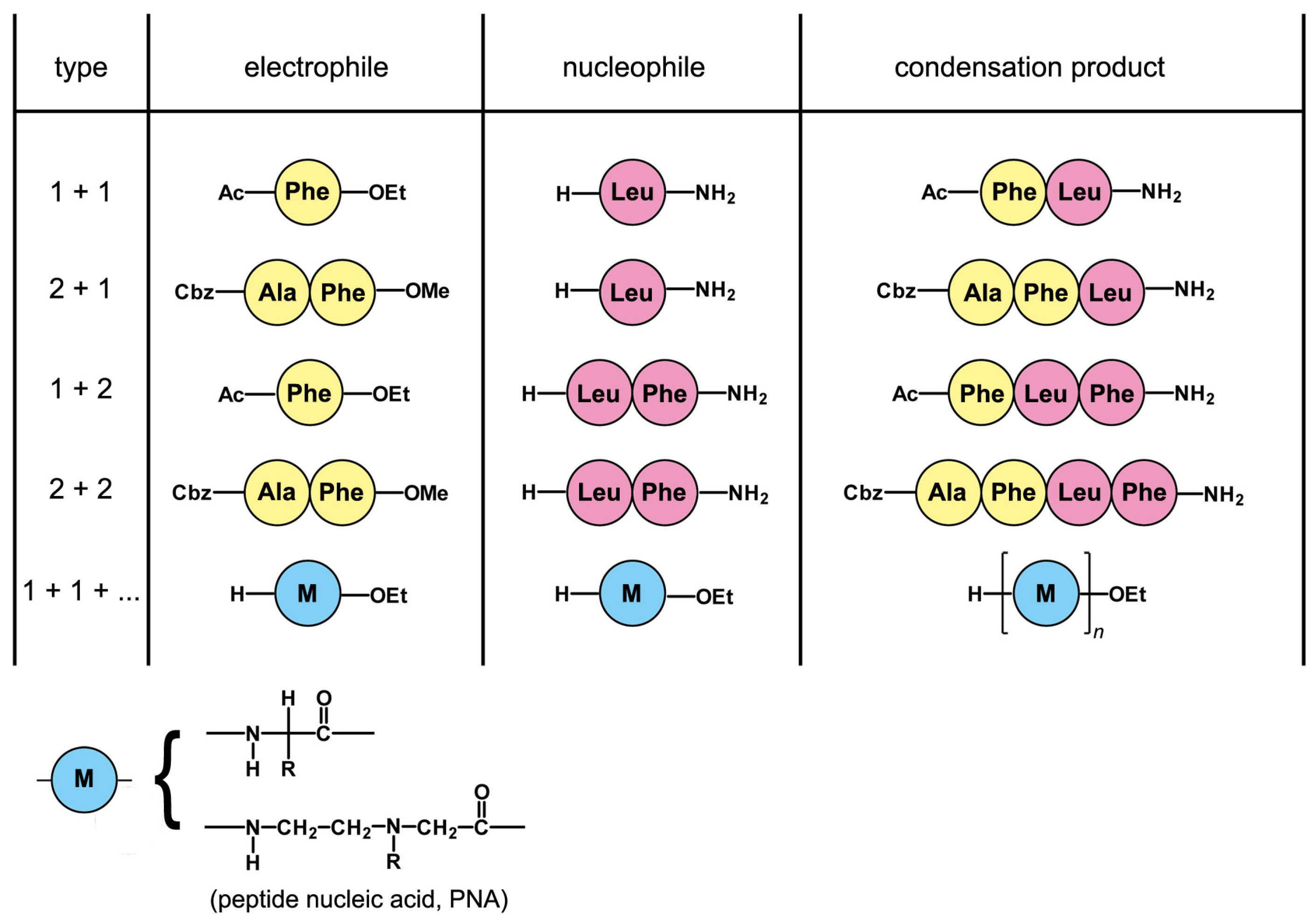
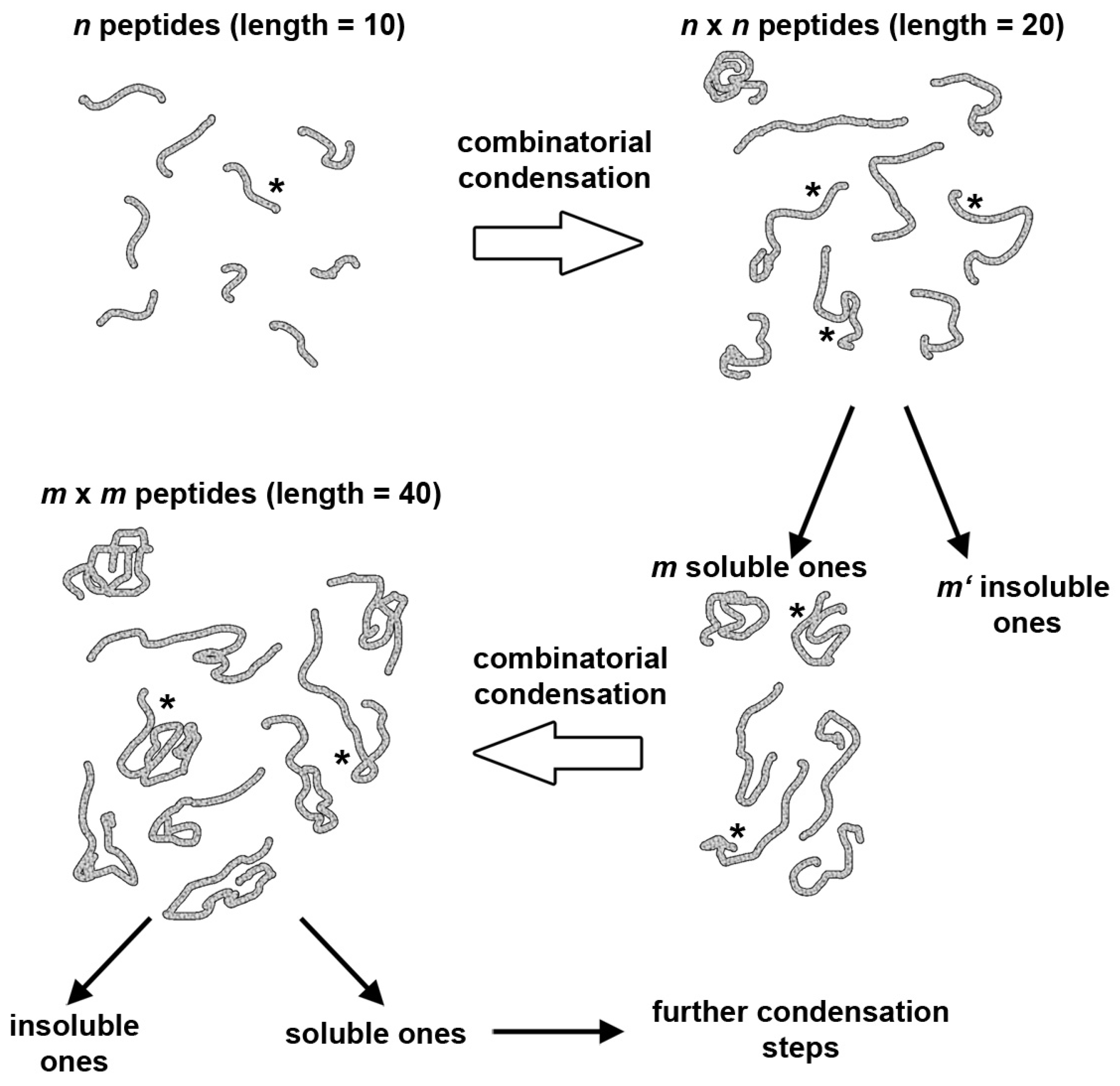
5. A Peptide “Fragment Condensation” Scenario
6. Other Potential Functions of Small Peptides
7. Looking for Short Random Primordial Peptides with Catalytic Functions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lambert, J.F. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Orgel, L.E. Prebiotic peptide-formation in the solid state. III. Condensation reactions of glycine in solid state mixtures containing inorganic polyphosphates. J. Mol. Evol. 1975, 6, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Chittenden, G.J.F.; Schwartz, A.W. Prebiotic photosynthetic reactions. Biosystems 1981, 14, 15–32. [Google Scholar] [CrossRef]
- Cleaves, H.J.; Aubrey, A.D.; Bada, J.L. An evaluation of the critical parameters for abiotic peptide synthesis in submarine hydrothermal systems. Orig. Life Evol. Biosph. 2009, 39, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.D.; Whitfield, J.N. Prebiotic chemistry: A bioorganic perspective. Tetrahedron 1997, 53, 11493–11527. [Google Scholar] [CrossRef]
- DeRose, V.J. Two decades of RNA catalysis. Chem. Biol. 2002, 9, 961–969. [Google Scholar] [CrossRef]
- Schwartz, A.W. Speculation on the RNA precursor problem. J. Theor. Biol. 1997, 187, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Szathmáry, E. The origin of the genetic code: Amino acids as cofactors in an RNA world. Trends Genet. 1999, 15, 223–229. [Google Scholar] [CrossRef]
- Plankensteiner, K.; Reiner, H.; Rode, B.M. Prebiotic Chemistry: The Amino Acid and Peptide World. Curr. Org. Chem. 2005, 9, 1107–1114. [Google Scholar] [CrossRef]
- Li, L.; Francklyn, C.; Carter, C.W. Aminoacylating urzymes challenge the RNA world hypothesis. J. Biol. Chem. 2013, 288, 26856–26863. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.T.; Zhou, M.; Burton, A.S.; Glavin, D.P.; Dworkin, J.P.; Krishnamurthy, R.; Fernández, F.M.; Bada, J.L. A plausible simultaneous synthesis of amino acids and simple peptides on the primordial Earth. Angew. Chem. Int. Ed. Engl. 2014, 53, 8132–8136. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Origin of life: The RNA world. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Kunin, V. A system of two polymerases—A model for the origin of life. Orig. Life Evol. Biosph. 2000, 30, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Van der Gulik, P.T.S.; Speijer, D. How amino acids and peptides shaped the RNA world. Life 2015, 5, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic systems chemistry: New perspectives for the origins of life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Gorlero, M.; Wieczorek, R.; Adamala, K.; Giorgi, A.; Schininà, M.E.; Stano, P.; Luisi, P.L. Ser-His catalyzes the formation of peptides and PNAs. FEBS Lett. 2009, 583, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, R.; Dörr, M.; Chotera, A.; Luisi, P.L.; Monnard, P.A. Formation of RNA phosphodiester bond by histidine-containing dipeptides. Chembiochem 2013, 14, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Adamala, K.; Anella, F.; Wieczorek, R.; Stano, P.; Chiarabelli, C.; Luisi, P.L. Open questions in origin of life: Experimental studies on the origin of nucleic acids and proteins with specific and functional sequences by a chemical synthetic biology approach. Comput. Struct. Biotechnol. J. 2014, 9, e201402004. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Y.; Hatfield, S.; Wan, R.; Zhu, Q.; Li, X.; McMills, M.; Ma, Y.; Li, J.; Brown, K.L.; et al. Dipeptide seryl-histidine and related oligopeptides cleave DNA, protein, and a carboxyl ester. Bioorg. Med. Chem. 2000, 8, 2675–2680. [Google Scholar] [CrossRef]
- Chaiken, I.M.; Komoriya, A.; Ohno, M.; Widmer, F. Use of enzymes in peptide synthesis. Appl. Biochem. Biotechnol. 1982, 7, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Fruton, J.S. Proteinase-catalyzed synthesis of peptide bonds. Adv. Enzymol. Relat. Areas Mol. Biol. 1982, 53, 239–306. [Google Scholar] [PubMed]
- Jakubke, H.D.; Kuhl, P.; Könnecke, A. Basic Principles of Protease-Catalyzed Peptide Bond Formation. Angew. Chem. Int. Ed. Engl. 1985, 24, 85–93. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels-Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Duncan, K.; Ulijn, R. Short peptides in minimalistic biocatalyst design. Biocatalysis 2015, 1, 67–81. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- Pizzarello, S. The Chemistry of Life’s Origin: A Carbonaceous Meteorite Perspective. Acc. Chem. Res. 2006, 39, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Doi, N.; Kakukawa, K.; Oishi, Y.; Yanagawa, H. High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Protein Eng. Des. Sel. 2005, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Zaia, D.A.M.; Zaia, C.T.B.V.; Santana, H.D. Which Amino Acids Should Be Used in Prebiotic Chemistry Studies? Orig. Life Evol. Biosph. 2008, 38, 469–488. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.D.; Storrie-Lombardi, M.C. Biochemical constraints in a protobiotic earth devoid of basic amino acids: The “BAA(-) world”. Astrobiology 2010, 10, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.M.; Blaber, M. Protein design at the interface of the pre-biotic and biotic worlds. Arch. Biochem. Biophys. 2012, 526, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Wolman, Y.; Haverland, W.J.; Miller, S.L. Nonprotein amino acids from spark discharges and their comparison with the murchison meteorite amino acids. Proc. Natl. Acad. Sci. USA 1972, 69, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Lerner, N.R.; Peterson, E.; Chang, S. The Strecker synthesis as a source of amino acids in carbonaceous chondrites: Deuterium retention during synthesis. Geochim. Cosmochim. Acta 1993, 57, 4713–4723. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. α-Hydroxy and α-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.M.; Lee, J.; Blaber, M. Simplified protein design biased for prebiotic amino acids yields a foldable, halophilic protein. Proc. Natl. Acad. Sci. USA 2013, 110, 2135–2139. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Yang, L.; Miller, S.L.; Oró, J. Prebiotic synthesis of imidazole-4-acetaldehyde and histidine. Orig. Life Evol. Biosph. 1987, 17, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Orgel, L.E. Studies on prebiotic synthesis. I. Aminomalononitrile and 4-amino-5-cyanoimidazole. J. Am. Chem. Soc. 1966, 88, 3829–3831. [Google Scholar] [CrossRef] [PubMed]
- Oró, J.; Basile, B.; Cortes, S.; Shen, C.; Yamrom, T. The prebiotic synthesis and catalytic role of imidazoles and other condensing agents. Orig. Life 1984, 14, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Salazar, A.; Tan, G.; Stockton, A.; Fani, R.; Becerra, A.; Lazcano, A. Can an Imidazole Be Formed from an Alanyl-Seryl-Glycine Tripeptide under Possible Prebiotic Conditions? Orig. Life Evol. Biosph. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Yang, L.; Miller, S.L.; Oro, J. Prebiotic synthesis of histidine. J. Mol. Evol. 1990, 31, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Weber, A.L. Prebiotic Amino Acids as Asymmetric Catalysts. Science 2004, 303, 1151. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Ibrahem, I.; Dziedzic, P.; Sundén, H.; Córdova, A. Small peptides as modular catalysts for the direct asymmetric aldol reaction: Ancient peptides with aldolase enzyme activity. Chem. Commun. 2005, 37, 4946–4948. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.B. Free energies and equilibria of peptide bond hydrolysis and formation. Biopolymers 1998, 45, 351–353. [Google Scholar] [CrossRef]
- Zepik, H.; Shavit, E.; Tang, M.; Jensen, T.R.; Kjaer, K.; Bolbach, G.; Leiserowitz, L.; Weissbuch, I.; Lahav, M. Chiral amplification of oligopeptides in two-dimensional crystalline self-assemblies on water. Science 2002, 295, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Eliash, R.; Weissbuch, I.; Weygand, M.J.; Kjaer, K.; Leiserowitz, L.; Lahav, M. Structure and Reactivity in Langmuir Films of Amphiphilic Alkyl and Thio-alkyl Esters of alpha-Amino Acids at the Air/Water Interface. J. Phys. Chem. B 2004, 108, 7228–7240. [Google Scholar] [CrossRef]
- Suwannachot, Y.; Rode, B.M. Mutual amino acid catalysis in salt-induced peptide formation supports this mechanism’s role in prebiotic peptide evolution. Orig. Life Evol. Biosph. 1999, 29, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fitz, D.; Rode, B.M. Isoleucine as a possible bridge between exogenous delivery and terrestrial enhancement of homochirality. Amino Acids 2013, 44, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Fiser, B.; Jójárt, B.; Szori, M.; Lendvay, G.; Csizmadia, I.G.; Viskolcz, B. Glutathione as a prebiotic answer to alpha-peptide based life. J. Phys. Chem. B 2015, 119, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, M.A.; Xu, Y.; Bebie, J. Energetics and kinetics of the prebiotic synthesis of simple organic acids and amino acids with the FeS-H2S/FeS2 redox couple as reductant. Orig. Life Evol. Biosph. 1999, 29, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Jauker, M.; Griesser, H.; Richert, C. Spontaneous Formation of RNA Strands, Peptidyl RNA, and Cofactors. Angew. Chem. Int. Ed. Engl. 2015, 54, 14564–14569. [Google Scholar] [CrossRef] [PubMed]
- Leman, L.J.; Huang, Z.Z.; Ghadiri, M.R. Peptide Bond Formation in Water Mediated by Carbon Disulfide. Astrobiology 2015, 15, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ehler, K.W.; Orgel, L.E. N,N′-carbonyldiimidazole-induced peptide formation in aqueous solution. Biochim. Biophys. Acta 1976, 434, 233–243. [Google Scholar] [CrossRef]
- Taillades, J.; Collet, H.; Garrel, L.; Beuzelin, I.; Boiteau, L.; Choukroun, H.; Commeyras, A. N-Carbamoyl Amino Acids Solid-Gas Nitrosation by NO/NOx: A New Route to Oligopeptides via α-Amino Acid N-Carboxyanhydride. Prebiotic Implications. J. Mol. Evol. 1999, 48, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Danger, G.; Boiteau, L.; Cottet, H.; Pascal, R. The peptide formation mediated by cyanate revisited. N-carboxyanhydrides as accessible intermediates in the decomposition of N-carbamoylamino acids. J. Am. Chem. Soc. 2006, 128, 7412–7413. [Google Scholar] [PubMed]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Hitz, T.; Luisi, P.L. Liposome-assisted selective polycondensation of alpha-amino acids and peptides. Biopolymers 2000, 55, 381–390. [Google Scholar] [CrossRef]
- Hitz, T.; Blocher, M.; Walde, P.; Luisi, P.L. Stereoselectivity Aspects in the Condensation of Racemic NCA-Amino Acids in the Presence and Absence of Liposomes. Macromolecules 2001, 34, 2443–2449. [Google Scholar] [CrossRef]
- Murillo-Sánchez, S.; Beaufils, D.; Mañas, J.M.G.; Pascal, R.; Ruiz-Mirazo, K. Fatty acids’ double role in the prebiotic formation of a hydrophobic dipeptide. Chem. Sci. 2016, 7, 3406–3413. [Google Scholar] [CrossRef]
- Kauffman, S. The Origins of Order: Self Organization and Selection in Evolution; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Lee, D.H.; Severin, K.; Ghadiri, M.R. Autocatalytic networks: The transition from molecular self-replication to molecular ecosystems. Curr. Opin. Chem. Biol. 1997, 1, 491–496. [Google Scholar] [CrossRef]
- Saghatelian, A.; Yokobayashi, Y.; Soltani, K.; Ghadiri, M.R. A chiroselective peptide replicator. Nature 2001, 409, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Ghosh, I.; Zutshi, R.; Chmielewski, J. A pH-Modulated, Self-Replicating Peptide. J. Am. Chem. Soc. 1997, 119, 10559–10560. [Google Scholar] [CrossRef]
- Issac, R.; Ham, Y.W.; Chmielewski, J. The design of self-replicating helical peptides. Curr. Opin. Struct. Biol. 2001, 11, 458–463. [Google Scholar] [CrossRef]
- Rubinov, B.; Wagner, N.; Rapaport, H.; Ashkenasy, G. Self-replicating amphiphilic beta-sheet peptides. Angew. Chem. Int. Ed. Engl. 2009, 48, 6683–6686. [Google Scholar] [CrossRef] [PubMed]
- Tena-Solsona, M.; Nanda, J.; Díaz-Oltra, S.; Chotera, A.; Ashkenasy, G.; Escuder, B. Emergent Catalytic Behavior of Self-Assembled Low Molecular Weight Peptide-Based Aggregates and Hydrogels. Chem. Eur. J. 2016, 22, 6687–6694. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L.; Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W H Freeman: New York, NY, USA, 2002. [Google Scholar]
- Buller, A.R.; Townsend, C.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.M.; Cravatt, B.F. Activity-based proteomics of enzyme superfamilies: Serine hydrolases as a case study. J. Biol. Chem. 2010, 285, 11051–11055. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wan, R.; Liu, H.; Cheng, C.M.; Zhao, Y.F. Cleavage of BSA by a dipeptide seryl-histidine. Lett. Pept. Sci. 2000, 7, 325–329. [Google Scholar] [CrossRef]
- Li, Y.; Hatfield, S.; Li, J.; McMills, M.; Zhao, Y.; Chen, X. Seryl-histidine as an alternative DNA nicking agent in nick translation yields superior DNA probes and hybridizations. Bioorg. Med. Chem. 2002, 10, 667–673. [Google Scholar] [CrossRef]
- Du, H.; Wang, Y.; Yang, L.; Luo, W.; Xia, N.; Zhaoh, Y. Appraisal of green fluorescent protein as a model substrate for seryl-histidine dipeptide cleaving agent. Lett. Pept. Sci. 2002, 9, 5–10. [Google Scholar] [CrossRef]
- Sun, M.; Zhu, C.; Tan, B.; Jin, Y.; Zhao, Y. Molecular modeling of diastereoisomeric aggregates of l/d ser/histamine amide with 5’-TpTpdC-3’. J. Mol. Model. 2003, 9, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Ma, Y.; Ji, S.; Liu, H.; Zhao, Y. Molecular modeling on DNA cleavage activity of seryl-histidine and related dipeptide. Bioorg. Med. Chem. Lett. 2004, 14, 3711–3714. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, J.; Liu, X.H.; Zhao, Y.F. Molecular interaction modeling of Ser-His dipeptide and buffers. J. Mol. Struct. Theochem 2004, 668, 47–49. [Google Scholar] [CrossRef]
- Zeng, Q.; Yin, Q.; Zhao, Y. The study on the interaction between seryl-histidine dipeptide and proteins by circular dichroism and molecular modeling. Bioorg. Med. Chem. 2005, 13, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, X.; Sun, M.; Wan, R.; Zhu, C.; Li, Y.; Zhao, Y. DNA cleavage function of seryl-histidine dipeptide and its application. Amino Acids 2008, 35, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hou, J.B.; Liu, X.X.; Miao, F.M.; Zhao, Y.F. Structural studies of L-Seryl-L-histidine dipeptide by means of molecular modeling, DFT and 1H NMR spectroscopy. J. Struct. Chem. 2009, 50, 835–840. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y.; Liu, X.; Lin, M.; Lin, D.; Zhao, Y. Evaluation of non-covalent interaction between Seryl-Histidine dipeptide and cyclophilin A using NMR and molecular modeling. Sci. China Chem. 2010, 53, 1987–1993. [Google Scholar] [CrossRef]
- Bongers, J.; Felix, A.; Heimer, E. Enzymatic semisynthesis of Growth Ormone-releasing factor and potent analogs. In Peptides: Design, Synthesis, and Biological Activity; Basava, C., Anantharamaiah, G.M., Eds.; Birkhäuser Boston: Boston, MA, USA, 1994; pp. 59–68. [Google Scholar]
- Schellenberger, V.; Jakubke, H.D. Protease-Catalyzed Kinetically Controlled Peptide Synthesis. Angew. Chem. Int. Ed. Engl. 1991, 30, 1437–1449. [Google Scholar] [CrossRef]
- Jencks, W. Free energies of hydrolysis and decarboxylation. In Handbook of Biochemistry and Molecular Biology, 3rd ed.; Fasman, G.D., Ed.; CRC Press: Cleveland, OH, USA, 1976; Volume I, pp. 296–304. [Google Scholar]
- Schuster, M.; Aaviksaar, A.; Jakubke, H.D. Enzyme-catalyzed peptide synthesis in ice. Tetrahedron 1990, 46, 8093–8102. [Google Scholar] [CrossRef]
- Bordusa, F. Proteases in organic synthesis. Chem. Rev. 2002, 102, 4817–4868. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [PubMed]
- Gorlero, M.; (University of RomaTre, Rome, Italy); Stano, P.; (University of Salento, Lecce, Italy). Personal Communication, 2008.
- Adamala, K.; Szostak, J.W. Competition between model protocells driven by an encapsulated catalyst. Nat. Chem. 2013, 5, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, R. On prebiotic ecology, supramolecular selection and autopoiesis. Orig. Life Evol. Biosph. 2012, 42, 445–450; discussion 451–452. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Bagnacani, V.; Corradini, R.; Nielsen, P.E. Toward peptide nucleic acid (PNA) directed peptide translation using ester based aminoacyl transfer. ACS Chem. Biol. 2014, 9, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Kanavarioti, A.; Monnard, P.A.; Deamer, D.W. Eutectic phases in ice facilitate nonenzymatic nucleic acid synthesis. Astrobiology 2001, 1, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.A.; Ziock, H. Eutectic phase in water-ice: A self-assembled environment conducive to metal-catalyzed non-enzymatic RNA polymerization. Chem. Biodivers. 2008, 5, 1521–1539. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Leung, I.; Cormier, M.; Sendelbeck, S.; Muchnik, A. Buffered Drug Formulations for Transdermal Electrotransport Delivery. WO Patent App. PCT/US1998/023,411, 20 May 1999. [Google Scholar]
- Ekici, O.D.; Paetzel, M.; Dalbey, R.E. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008, 17, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Frankel, A.D. Structural variety of arginine-rich RNA-binding peptides. Proc. Natl. Acad. Sci. USA 1995, 92, 5282–5286. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.E.; Kent, S.B. Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 2000, 69, 923–960. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, S.; Datta, D.; Roy, R.P. An enigmatic peptide ligation reaction: Protease-catalyzed oligomerization of a native protein segment in neat aqueous solution. Protein Sci. 2000, 9, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L. The Emergence of Life: From Chemical Origins to Synthetic Biology, 1st ed.; Cambridge University Press: Cambridge UK, 2006. [Google Scholar]
- Chessari, S.; Thomas, R.; Polticelli, F.; Luisi, P.L. The production of de novo folded proteins by a stepwise chain elongation: A model for prebiotic chemical evolution of macromolecular sequences. Chem. Biodivers. 2006, 3, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A. The discovery of catalytically active peptides through combinatorial chemistry. Curr. Opin. Chem. Biol. 2003, 7, 409–419. [Google Scholar] [CrossRef]
- Wei, Y.; Hecht, M.H. Enzyme-like proteins from an unselected library of designed amino acid sequences. Protein Eng. Des. Sel. 2004, 17, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ginalski, K.; Elofsson, A.; Fischer, D.; Rychlewski, L. 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 2003, 19, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Luisi, P.L.; Allegretti, M.; Pereira de Souza, T.; Steiniger, F.; Fahr, A.; Stano, P. Spontaneous protein crowding in liposomes: A new vista for the origin of cellular metabolism. Chembiochem 2010, 11, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Pereira de Souza, T.; Steiniger, F.; Stano, P.; Fahr, A.; Luisi, P.L. Spontaneous crowding of ribosomes and proteins inside vesicles: A possible mechanism for the origin of cell metabolism. Chembiochem 2011, 12, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Stano, P.; D’Aguanno, E.; Bolz, J.; Fahr, A.; Luisi, P.L. A remarkable self-organization process as the origin of primitive functional cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 13397–13400. [Google Scholar] [CrossRef] [PubMed]
- Somalinga, B.R.; Roy, R.P. Volume exclusion effect as a driving force for reverse proteolysis. Implications for polypeptide assemblage in a macromolecular crowded milieu. J. Biol. Chem. 2002, 277, 43253–43261. [Google Scholar] [CrossRef] [PubMed]
- Grochmal, A.; Prout, L.; Makin-Taylor, R.; Prohens, R.; Tomas, S. Modulation of Reactivity in the Cavity of Liposomes Promotes the Formation of Peptide Bonds. J. Am. Chem. Soc. 2015, 137, 12269–12275. [Google Scholar] [CrossRef] [PubMed]
- Raine, D.J.; Norris, V. Lipid domain boundaries as prebiotic catalysts of peptide bond formation. J. Theor. Biol. 2007, 246, 176–185. [Google Scholar] [CrossRef] [PubMed]
- DeRouchey, J.; Hoover, B.; Rau, D.C. A comparison of DNA compaction by arginine and lysine peptides: A physical basis for arginine rich protamines. Biochemistry 2013, 52, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Balhorn, R.; Brewer, L.; Corzett, M. DNA condensation by protamine and arginine-rich peptides: Analysis of toroid stability using single DNA molecules. Mol. Reprod. Dev. 2000, 56, 230–234. [Google Scholar] [CrossRef]
- Bártová, E.; Krejcí, J.; Harnicarová, A.; Galiová, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Mascotti, D.P.; Lohman, T.M. Thermodynamics of oligoarginines binding to RNA and DNA. Biochemistry 1997, 36, 7272–7279. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Garde, S.; García, A.E. Role of backbone hydration and salt-bridge formation in stability of alpha-helix in solution. Biophys. J. 2003, 85, 3187–3193. [Google Scholar] [CrossRef]
- Liu, R.; Orgel, L.E. Polymerization on the rocks: Beta-amino acids and arginine. Orig. Life Evol. Biosph. 1998, 28, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Ren, J.; Xiang, J.; Yan, Q.; Xie, Y.; Wang, K.; Lai, P.; Bai, Y. 6-Membered ring intermediates in polymerization of N-carboxyanhydride-L-alpha-arginine in H2O. Sci. China Ser. B Chem. 2009, 52, 1220–1226. [Google Scholar] [CrossRef]
- Szostak, J.W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012, 3, 2. [Google Scholar] [CrossRef]
- Kamat, N.P.; Tobé, S.; Hill, I.T.; Szostak, J.W. Electrostatic Localization of RNA to Protocell Membranes by Cationic Hydrophobic Peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 11735–11739. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.C.; Deamer, D.W. Permeability of lipid bilayers to amino acids and phosphate. Biochim. Biophys. Acta 1992, 1111, 171–177. [Google Scholar] [CrossRef]
- Deamer, D.W.; Dworkin, J.P. Chemistry and Physics of Primitive Membranes. In Prebiotic Chemistry; Number 259 in Topics in Current Chemistry; Walde, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–27. [Google Scholar] [CrossRef]
- Wilson, M.A.; Wei, C.; Pohorille, A. Towards co-evolution of membrane proteins and metabolism. Orig. Life Evol. Biosph. 2014, 44, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Lamy, C.; Lemoine, J.; Bouchu, D.; Goekjian, P.; Strazewski, P. Glutamate–Glycine and Histidine–Glycine Co-oligopeptides: Batch Co-oligomerization versus Pulsed Addition of N-Carboxyanhydrides. ChemBioChem 2008, 9, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Schwab, L.W.; Kloosterman, W.M.J.; Konieczny, J.; Loos, K. Papain Catalyzed (co)Oligomerization of α-Amino Acids. Polymers 2012, 4, 710–740. [Google Scholar] [CrossRef]
- Wolfenden, R.; Snider, M.J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.G.; Wolfenden, R. Catalytic proficiency: The unusual case of OMP decarboxylase. Annu. Rev. Biochem. 2002, 71, 847–885. [Google Scholar] [CrossRef] [PubMed]
- Chiarabelli, C.; Vrijbloed, J.W.; Thomas, R.M.; Luisi, P.L. Investigation of de novo totally random biosequences, Part I: A general method for in vitro selection of folded domains from a random polypeptide library displayed on phage. Chem. Biodivers. 2006, 3, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Chiarabelli, C.; Vrijbloed, J.W.; De Lucrezia, D.; Thomas, R.M.; Stano, P.; Polticelli, F.; Ottone, T.; Papa, E.; Luisi, P.L. Investigation of de novo totally random biosequences, Part II: On the folding frequency in a totally random library of de novo proteins obtained by phage display. Chem. Biodivers. 2006, 3, 840–859. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, S.; Schiffer, J.M.; Xiong, H.; Babik, J.M.; Hecht, M.H. Protein design by binary patterning of polar and nonpolar amino acids. Science 1993, 262, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.R.; Lumb, K.J.; Sauer, R.T. Cooperatively folded proteins in random sequence libraries. Nat. Struct. Biol. 1995, 2, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stöhr, J.; Smith, T.A.; Hu, X.; DeGrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tena-Solsona, M.; Miravet, J.F.; Escuder, B. Towards Supramolecular Catalysis with Small Self-assembled Peptides. Isr. J. Chem. 2015, 55, 711–723. [Google Scholar] [CrossRef]
- Authors, M. Workshop OQOL’09: Open Questions on the Origins of Life 2009. Orig. Life Evol. Biosph. 2010, 40, 347–497. [Google Scholar]
- MacDonald, M.J.; Lavis, L.D.; Hilvert, D.; Gellman, S.H. Evaluation of the Ser-His Dipeptide, a Putative Catalyst of Amide and Ester Hydrolysis. Org. Lett. 2016, 18, 3518–3521. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieczorek, R.; Adamala, K.; Gasperi, T.; Polticelli, F.; Stano, P. Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine. Life 2017, 7, 19. https://doi.org/10.3390/life7020019
Wieczorek R, Adamala K, Gasperi T, Polticelli F, Stano P. Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine. Life. 2017; 7(2):19. https://doi.org/10.3390/life7020019
Chicago/Turabian StyleWieczorek, Rafal, Katarzyna Adamala, Tecla Gasperi, Fabio Polticelli, and Pasquale Stano. 2017. "Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine" Life 7, no. 2: 19. https://doi.org/10.3390/life7020019
APA StyleWieczorek, R., Adamala, K., Gasperi, T., Polticelli, F., & Stano, P. (2017). Small and Random Peptides: An Unexplored Reservoir of Potentially Functional Primitive Organocatalysts. The Case of Seryl-Histidine. Life, 7(2), 19. https://doi.org/10.3390/life7020019