Evaluation of Chromosome Microarray Analysis in a Large Cohort of Females with Autism Spectrum Disorders: A Single Center Italian Study

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Methods
3. Procedure
3.1. Genetic Analysis
3.2. Statistical Analyses
4. Results
4.1. Chromosome Microarray Analysis (CMA)
4.2. Phenotypic Characterization
4.3. Statistical Analysis
5. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Statement
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Publication: Washington, DC, USA, 2013. [Google Scholar]
- Muhle, R.A.; Reed, H.E.; Stratigos, K.A.; Weele, J.V.-V. The Emerging Clinical Neuroscience of Autism Spectrum Disorder: A Review. JAMA Psychiatry 2018, 75, 514–523. [Google Scholar] [CrossRef]
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; et al. Environmental risk factors and biomarkers for autism spectrum disorder: An umbrella review of the evidence. Lancet Psychiatry 2019, 6, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Narzisi, A.; Posada, M.; Barbieri, F.; Chericoni, N.; Ciuffolini, D.; Pinzino, M.; Romano, R.; Scattoni, M.L.; Tancredi, R.; Calderoni, S.; et al. Prevalence of Autism Spectrum Disorder in a large Italian catchment area: A school-based population study within the ASDEU project. Epidemiol. Psychiatr. Sci. 2018, 29, e5. [Google Scholar] [CrossRef]
- Kanner, L. Autistic disturbances of affective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- Asperger, H. Die “Autistischen Psychopathen” im Kindesalter. Arch. Psychiatr. Nervenkr. 1944, 117, 76–136. [Google Scholar] [CrossRef]
- Wing, L. Sex ratios in early childhood autism and related conditions. Psychiatry Res. 1981, 5, 129–137. [Google Scholar] [CrossRef]
- Lord, C.; Schopler, E. Neurobiological Implications of Sex Differences in Autism. In Neurobiological Issues in Autism; Schopler, E., Mesibov, G., Eds.; Plenum Press: New York, NY, USA, 1987; pp. 191–211. [Google Scholar]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef]
- McCarthy, M.M.; Wright, C.L. Convergence of Sex Differences and the Neuroimmune System in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 402–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werling, D.M.; Parikshak, N.N.; Geschwind, D.H. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 2016, 7, 10717. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Desachy, G.; Croen, L.A.; Torres, A.R.; Kharrazi, M.; Delorenze, G.N.; Windham, G.C.; Yoshida, C.K.; Weiss, L.A. Increased female autosomal burden of rare copy number variants in human populations and in autism families. Mol. Psychiatry 2015, 20, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Mitra, I.; Tsang, K.; Ladd-Acosta, C.; Croen, L.A.; Aldinger, K.A.; Hendren, R.L.; Traglia, M.; Lavillaureix, A.; Zaitlen, N.; Oldham, M.C.; et al. Pleiotropic Mechanisms Indicated for Sex Differences in Autism. PLoS Genet. 2016, 12, e1006425. [Google Scholar] [CrossRef]
- Van Wijngaarden-Cremers, P.J.M.; van Eeten, E.; Groen, W.B.; Van Deurzen, P.A.; Oosterling, I.J.; Van der Gaag, R.J. Gender and age differences in the core triad of impairments in autism spectrum disorders: A systematic review and meta-analysis. J. Autism Dev. Disord. 2014, 44, 627–635. [Google Scholar] [CrossRef]
- Banach, R.; Thompson, A.; Szatmari, P.; Goldberg, J.; Tuff, L.; Zwaigenbaum, L.; Mahoney, W. Brief Report: Relationship between non-verbal IQ and gender in autism. J. Autism Dev. Disord. 2009, 39, 188–193. [Google Scholar] [CrossRef]
- Carter, A.S.; Black, D.O.; Tewani, S.; Connolly, C.E.; Kadlec, M.B.; Tager-Flusberg, H. Sex differences in toddlers with autism spectrum disorders. J. Autism Dev. Disord. 2007, 37, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, M.; Bölte, S.; Poustka, F. Autism spectrum disorders: Sex differences in autistic behaviour domains and coexisting psychopathology. Dev. Med. Child Neurol. 2007, 49, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Duvekot, J.; van der Ende, J.; Verhulst, F.C.; Slappendel, G.; van Daalen, E.; Maras, A.; Greaves-Lord, K. Factors influencing the probability of a diagnosis of autism spectrum disorder in girls versus boys. Autism 2017, 21, 646–658. [Google Scholar] [CrossRef]
- Hartley, S.L.; Sikora, D.M. Sex differences in autism spectrum disorder: An examination of developmental functioning, autistic symptoms, and coexisting behavior problems in toddlers. J. Autism Dev. Disord. 2009, 39, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Tillmann, J.; Ashwood, K.; Absoud, M.; Bölte, S.; Bonnet-Brilhault, F.; Buitelaar, J.K.; Calderoni, S.; Calvo, R.; Canal-Bedia, R.; Canitano, R.; et al. Evaluating Sex and Age Differences in ADI-R and ADOS Scores in a Large European Multi-site Sample of Individuals with Autism Spectrum Disorder. J. Autism Dev. Disord. 2018, 48, 2490–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, G.W.; Gillberg, C.; Miniscalco, C. Pre-school children with suspected autism spectrum disorders: Do girls and boys have the same profiles? Res. Dev. Disabil. 2013, 34, 413–422. [Google Scholar] [CrossRef]
- Postorino, V.; Fatta, L.M.; De Peppo, L.; Giovagnoli, G.; Armando, M.; Vicari, S.; Mazzone, L. Longitudinal comparison between male and female preschool children with autism spectrum disorder. J. Autism Dev. Disord. 2015, 45, 2046–2055. [Google Scholar] [CrossRef] [PubMed]
- Messinger, D.S.; Young, G.S.; Webb, S.J.; Ozonoff, S.; Bryson, S.E.; Carter, A.; Carver, L.; Charman, T.; Chawarska, K.; Curtin, S.; et al. Early sex differences are not autism-specific: A Baby Siblings Research Consortium (BSRC) study. Mol. Autism 2015, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; Magalhaes, T.; Conroy, J.M.; Regan, R.; Shah, N.; Anney, R.; Shields, D.C.; Abrahams, B.S.; Almeida, J.; Bacchelli, E.; et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum. Genet. 2012, 131, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Zwaigenbaum, L.; Bryson, S.E.; Szatmari, P.; Brian, J.; Smith, I.M.; Roberts, W.; Vaillancourt, T.; Roncadin, C. Sex differences in children with autism spectrum disorder identified within a high-risk infant cohort. J. Autism Dev. Disord. 2012, 42, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Ben-Itzchak, E.; Ben-Shachar, S.; Zachor, D.A. Specific neurological phenotypes in autism spectrum disorders are associated with sex representation. Autism Res. 2013, 6, 596–604. [Google Scholar] [CrossRef]
- Amiet, C.; Gourfinkel-An, I.; Bouzamondo, A.; Tordjman, S.; Baulac, M.; Lechat, P.; Mottron, L.; Cohen, D. Epilepsy in autism is associated with intellectual disability and gender: Evidence from a meta-analysis. Biol. Psychiatry 2008, 64, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Shaw, C.A.; Yu, W.; Li, J.; Ou, Z.; Patel, A.; Yatsenko, S.A.; Cooper, M.L.; Furman, P.; Stankiewicz, P.; et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet. Med. 2005, 7, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Committee on Bioethics, Committee on Genetics, and the American College of Medical Genetics and Genomics Social, Ethical, and Legal Issues Committee. Ethical and Policy Issues in Genetic Testing and Screening of Children. Pediatrics 2013, 131, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Manning, M.; Hudgins, L. Professional Practice and Guidelines Committee Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [Green Version]
- Volkmar, F.; Siegel, M.; Woodbury-Smith, M.; King, B.; McCracken, J.; State, M.; American Academy of Child and Adolescent Psychiatry (AACAP) Committee on Quality Issues (CQI). Practice parameter for the assessment and treatment of children and adolescents with autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 237–257. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; Pan, C.; Chen, L.; Hadd, A.G.; Russell, R. A dual-mode single-molecule fluorescence assay for the detection of expanded CGG repeats in Fragile X syndrome. Mol. Biotechnol. 2013, 53, 19–28. [Google Scholar] [CrossRef]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.C.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. JAMA 2015, 314, 895–903. [Google Scholar] [CrossRef]
- Miles, J.H.; Takahashi, T.N.; Bagby, S.; Sahota, P.K.; Vaslow, D.F.; Wang, C.H.; Hillman, R.E.; Farmer, J.E. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am. J. Med. Genet. Part A 2005, 135, 171–180. [Google Scholar] [CrossRef]
- Griffiths, R. The Griffiths Mental Developmental Scales, Revised. Henley: Association for Research in Infant and Child Development; Test Agency: Oxford, UK, 1996. [Google Scholar]
- Wechsler, D. WPPSI-III Wechsler Preschool and Primary Scale of Intelligence—III. Adattamento Italiano a cura di G. Sannio Fancello e C. Cianchetti; Giunti O.S.: Firenze, Italy, 2008. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children—Fourth Edition (WISC-IV); The Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Roid, G.H.; Miller, L.J. The Leiter International Performance Scale-Revised Edition; Psychological Assessment Resources: Lutz, FL, USA, 1997. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S. ADOS-2 Autism Diagnostic Observation Schedule, 2nd ed.; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Gotham, K.; Pickles, A.; Lord, C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord. 2009, 39, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Esler, A.N.; Bal, V.H.; Guthrie, W.; Wetherby, A.; Weismer, S.E.; Lord, C. The Autism Diagnostic Observation Schedule, Toddler Module: Standardized Severity Scores. J. Autism Dev. Disord. 2015, 45, 2704–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Database of Genomic Variants (DGV). Available online: http://dgv.tcag.ca/dgv/app/home (accessed on 26 July 2020).
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man (OMIM) Database. Available online: https://www.omim.org (accessed on 26 July 2020).
- Simons Foundation Autism Research Initiative (SFARI). Gene Database. Available online: https://gene.sfari.org/database/human-gene (accessed on 26 July 2020).
- Autism KnowledgeBase Version 2.0 (Autism KB 2.0). Database. Available online: http://db.cbi.pku.edu.cn/autismkb_v2/index.php (accessed on 26 July 2020).
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- ToppGene Suite. Available online: https://toppgene.cchmc.org (accessed on 31 August 2020).
- Lal, D.; Pernhorst, K.; Klein, K.M.; Reif, P.; Tozzi, R.; Toliat, M.R.; Winterer, G.; Neubauer, B.; Nürnberg, P.; Rosenow, F.; et al. Extending the phenotypic spectrum of RBFOX1 deletions: Sporadic focal epilepsy. Epilepsia 2015, 56, e129–e133. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Tucci, A.; Scuvera, G.; Estienne, M.; Esposito, S.; Milani, D. 16p13 microduplication without CREBBP involvement: Moving toward a phenotype delineation. Eur. J. Med. Genet. 2017, 60, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Xie, H.M.; Perin, J.C.; Takahashi, N.; Murphy, K.; Wenocur, A.S.; D’Arcy, M.; O’Hara, R.J.; Goldmuntz, E.; Grice, D.E.; et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 2012, 17, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizuka, K.; Kimura, H.; Wang, C.; Xing, J.; Kushima, I.; Arioka, Y.; Oya-Ito, T.; Uno, Y.; Okada, T.; Mori, D.; et al. Investigation of Rare Single-Nucleotide PCDH15 Variants in Schizophrenia and Autism Spectrum Disorders. PLoS ONE 2016, 11, e0153224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.L.; Merriman, B.; Cantor, R.M.; Geschwind, D.H.; Nelson, S.F. High density SNP association study of a major autism linkage region on chromosome 17. Hum. Mol. Genet. 2007, 16, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Pan, C. Role of metabotropic glutamate receptor 7 in autism spectrum disorders: A pilot study. Life Sci. 2013, 92, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Baldan, F.; Gnan, C.; Franzoni, A.; Ferino, L.; Allegri, L.; Passon, N.; Damante, G. Genomic Deletion Involving the IMMP2L Gene in Two Cases of Autism Spectrum Disorder. Cytogenet. Genome Res. 2018, 154, 196–200. [Google Scholar] [CrossRef]
- Ben Khelifa, H.; Soyah, N.; Ben-Abdallah-Bouhjar, I.; Gritly, R.; Sanlaville, D.; Elghezal, H.; Saad, A.; Mougou-Zerelli, S. Xp22.3 interstitial deletion: A recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene 2013, 527, 578–583. [Google Scholar] [CrossRef]
- Van Rijn, S.; Stockmann, L.; Borghgraef, M.; Bruining, H.; van Ravenswaaij-Arts, C.; Govaerts, L.; Hansson, K.; Swaab, H. The social behavioral phenotype in boys and girls with an extra X chromosome (Klinefelter syndrome and Trisomy X): A comparison with autism spectrum disorder. J. Autism Dev. Disord. 2014, 44, 310–320. [Google Scholar] [CrossRef]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef]
- Guo, H.; Peng, Y.; Hu, Z.; Li, Y.; Xun, G.; Ou, J.; Sun, L.; Xiong, Z.; Liu, Y.; Wang, T.; et al. Genome-wide copy number variation analysis in a Chinese autism spectrum disorder cohort. Sci. Rep. 2017, 7, 44155. [Google Scholar] [CrossRef] [Green Version]
- Dabell, M.P.; Rosenfeld, J.A.; Bader, P.; Escobar, L.F.; El-Khechen, D.; Vallee, S.E.; Dinulos, M.B.P.; Curry, C.; Fisher, J.; Tervo, R.; et al. Investigation of NRXN1 deletions: Clinical and molecular characterization. Am. J. Med. Genet. Part A 2013, 161A, 717–731. [Google Scholar] [CrossRef]
- Tropeano, M.; Howley, D.; Gazzellone, M.J.; Wilson, C.E.; Ahn, J.W.; Stavropoulos, D.J.; Murphy, C.M.; Eis, P.S.; Hatchwell, E.; Dobson, R.J.B.; et al. Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J. Med. Genet. 2016, 53, 536–547. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice and Guidelines Committee Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chen, H.-I.; Chien, W.-H.; Li, L.-H.; Wu, Y.-Y.; Chiu, Y.-N.; Tsai, W.-C.; Gau, S.S.-F. High resolution analysis of rare copy number variants in patients with autism spectrum disorder from Taiwan. Sci. Rep. 2017, 7, 11919. [Google Scholar] [CrossRef] [Green Version]
- Napoli, E.; Russo, S.; Casula, L.; Alesi, V.; Amendola, F.A.; Angioni, A.; Novelli, A.; Valeri, G.; Menghini, D.; Vicari, S. Array-CGH Analysis in a Cohort of Phenotypically Well-Characterized Individuals with “Essential” Autism Spectrum Disorders. J. Autism Dev. Disord. 2018, 48, 442–449. [Google Scholar] [CrossRef]
- Barone, R.; Gulisano, M.; Amore, R.; Domini, C.; Milana, M.C.; Giglio, S.; Madia, F.; Mattina, T.; Casabona, A.; Fichera, M.; et al. Clinical correlates in children with autism spectrum disorder and CNVs: Systematic investigation in a clinical setting. Int. J. Dev. Neurosci. 2020, 80, 276–286. [Google Scholar] [CrossRef]
- Bacchelli, E.; Cameli, C.; Viggiano, M.; Igliozzi, R.; Mancini, A.; Tancredi, R.; Battaglia, A.; Maestrini, E. An integrated analysis of rare CNV and exome variation in Autism Spectrum Disorder using the Infinium PsychArray. Sci. Rep. 2020, 10, 3198. [Google Scholar] [CrossRef] [Green Version]
- Jacquemont, S.; Coe, B.P.; Hersch, M.; Duyzend, M.H.; Krumm, N.; Bergmann, S.; Beckmann, J.S.; Rosenfeld, J.A.; Eichler, E.E. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 2014, 94, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Girirajan, S.; Rosenfeld, J.A.; Coe, B.P.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.A.; McConnell, J.S.; Angle, B.; Meschino, W.S.; et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.; Hermetz, K.E.; Weckselblatt, B.; Rudd, M.K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 2015, 96, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, E.; Wiggins, L.D.; Lee, L.-C. A Review of the Differences in Developmental, Psychiatric, and Medical Endophenotypes Between Males and Females with Autism Spectrum Disorder. J. Dev. Phys. Disabil. 2015, 27, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Charman, T.; Loth, E.; Tillmann, J.; Crawley, D.; Wooldridge, C.; Goyard, D.; Ahmad, J.; Auyeung, B.; Ambrosino, S.; Banaschewski, T.; et al. The EU-AIMS Longitudinal European Autism Project (LEAP): Clinical characterisation. Mol. Autism 2017, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, J.; Crossman, M.; Perrin, J.; Shui, A.; Kuhlthau, K. Sex differences in restricted repetitive behaviors and interests in children with autism spectrum disorder: An Autism Treatment Network study. Autism 2019, 23, 858–868. [Google Scholar] [CrossRef]
- Warrier, V.; Toro, R.; Won, H.; Leblond, C.S.; Cliquet, F.; Delorme, R.; De Witte, W.; Bralten, J.; Chakrabarti, B.; Børglum, A.D.; et al. Social and non-social autism symptoms and trait domains are genetically dissociable. Commun. Biol. 2019, 2, 328. [Google Scholar] [CrossRef] [Green Version]
- Uljarević, M.; Evans, D.W.; Alvares, G.A.; Whitehouse, A.J.O. Short report: Relationship between restricted and repetitive behaviours in children with autism spectrum disorder and their parents. Mol. Autism 2016, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Szatmari, P.; Liu, X.-Q.; Goldberg, J.; Zwaigenbaum, L.; Paterson, A.D.; Woodbury-Smith, M.; Georgiades, S.; Duku, E.; Thompson, A. Sex differences in repetitive stereotyped behaviors in autism: Implications for genetic liability. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 5–12. [Google Scholar] [CrossRef]
- Mazina, V.; Gerdts, J.; Trinh, S.; Ankenman, K.; Ward, T.; Dennis, M.Y.; Girirajan, S.; Eichler, E.E.; Bernier, R. Epigenetics of autism-related impairment: Copy number variation and maternal infection. J. Dev. Behav. Pediatr. 2015, 36, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Niarchou, M.; Chawner, S.J.R.A.; Doherty, J.L.; Maillard, A.M.; Jacquemont, S.; Chung, W.K.; Green-Snyder, L.; Bernier, R.A.; Goin-Kochel, R.P.; Hanson, E.; et al. Psychiatric disorders in children with 16p11.2 deletion and duplication. Transl. Psychiatry 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.V.M.; Jacobs, P.A.; Lachlan, K.; Wellesley, D.; Barnicoat, A.; Boyd, P.A.; Fryer, A.; Middlemiss, P.; Smithson, S.; Metcalfe, K.; et al. Autism, language and communication in children with sex chromosome trisomies. Arch. Dis. Child. 2011, 96, 954–959. [Google Scholar] [CrossRef] [Green Version]
- Fountain, M.D.; Oleson, D.S.; Rech, M.E.; Segebrecht, L.; Hunter, J.V.; McCarthy, J.M.; Lupo, P.J.; Holtgrewe, M.; Moran, R.; Rosenfeld, J.A.; et al. Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies. Genet. Med. 2019, 21, 1797–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyses-Oliveira, M.; Yadav, R.; Erdin, S.; Talkowski, M.E. New gene discoveries highlight functional convergence in autism and related neurodevelopmental disorders. Curr. Opin. Genet. Dev. 2020, 65, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: Contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID. | CNV Breakpoints | CNV Type | Size (bp) | Inheritance | Syndrome/Candidate Gene | CNV Class | Reference |
|---|---|---|---|---|---|---|---|
| P1 | 22q13.33 (50781138-51219009) | del | 437,871 | de novo | Phelan-McDermid syndrome | C | #MIM 606232 |
| Xp11.4 (38491539-38628756) | dup | 137,217 | mat | TSPAN7 | C | #MIM 300210 | |
| P2 | 14q32.13 (94817951-94883978) | del | 66,027 | - | - | N | - |
| P3 | 16p13.3 (6881091-7070689) | del | 189,598 | mat | RBFOX1 | C | [59] |
| 16p13.2 (9015110-10321593) | dup | 1,306,483 | de novo | USP7, GRIN2A | C | [60] | |
| P4 | 21q22.3 (45822805-46530451) | dup | 707,646 | mat | ADARB1, TRPM2, ITGB2, SUMO3 | C | [61] |
| P5 | 1q31.2 (191644543-191775583) | del | 131,040 | pat | - | N | - |
| P6 | 2q34.3 (214919902-215051057) | del | 131,155 | mat | - | N | - |
| P7 | 1q21.2 (147211160-147824207) | dup | 613,047 | pat | - | N | - |
| P8 | 17p11.2 (16822483-20193310) | del | 3,370,827 | de novo | Smith-Magenis syndrome | C | #MIM 182290 |
| P9 | 15q21.3 (56283008-56384604) | del | 101,596 | mat | NEDD4, RFX7 | C | [62] |
| P10 | 17q12 (34851537-36168104) | del | 1,316,567 | de novo | 17q12 deletion syndrome | C | #MIM 614527 |
| P11 | 10q21.1 (55616917-55791973) | del | 175,056 | mat | PCDH15 | C | [63] |
| P12 | 4q34.1 (172930618-173074943) | dup | 144,325 | pat | - | N | - |
| 4q34.2 (176984739-177190235) | dup | 205,496 | pat | - | N | - | |
| P13 | 17q12 (31953228-32922965) | dup | 969,737 | mat | ACCN1, TMEM132E | C | [64] |
| P14 | 3p12.1 (85615568-85672801) | del | 57,233 | pat | CADM2 | C | [32] |
| 3p26.1 (7353126-7403750) | del | 50,624 | pat | GRM7 | C | [65] | |
| P15 | 7q31.1 (110954950-111202026) | del | 247,076 | pat | IMMP2L | C | [66] |
| P16 | 2q23.3 (153898093-154164672) | del | 266,579 | pat | - | N | - |
| P17 | 5q23.3 (129687092-130006500) | del | 319,408 | mat | - | N | - |
| P18 | 15q11.2q13.1 (23669701-28525460) | dup | 4,825,759 | - | 15q11q13 duplication syndrome | C | #MIM 608636 |
| P19 | 2p16.1p15 (58566616-61546442) | del | 2,979,826 | de novo | 2p16.1p15 deletion syndrome | C | #MIM 612513 |
| P20 | 16p11.2 (29673954-30197341) | dup | 523,387 | - | 16p11.2 duplication syndrome | C | #MIM 614671 |
| P21 | 15q11.2q13.1 (23669701-28525460) | dup | 4,855,759 | mat | 15q11-q13 duplication syndrome | C | #MIM 608636 |
| P22 | 9p24.1 (7800020-8528849) | dup | 728,829 | - | PTPRD | C | [61] |
| Xp22.31 (6552712-8115153) | del | 1,562,441 | - | Xp22.31 deletion syndrome | C | [67] | |
| P23 | 2p16.3 (48915312-48979903) | del | 64,591 | pat | - | N | - |
| P24 | Xp22.33q28 (61529-155190083) | dup | 155,128,554 | de novo | 47, XXX | C | [68] |
| P25 | Xp21.1 (31893344-32289012) | dup | 395,668 | de novo | DMD | C | [69] |
| P26 | 8q24.3 (146053353-146174033 | dup | 120,680 | - | - | N | - |
| P27 | 2q12.2q12.3 (106929257-108403252) | dup | 1,473,995 | pat | ST6GAL2 | C | [70] |
| P28 | 22q11.21 (20754422-21440514) | dup | 686,092 | pat | 22q11.2 duplication syndrome | C | #MIM 608363 |
| P29 | 2p16.2 (50909765-51083469) | del | 173,704 | pat | NRXN1 | C | [71] |
| Xp22.33 (581803-920279) | dup | 338,476 | de novo | SHOX | C | [72] |
| Gene | Genomic Region (Participant ID) | Protein Function | SFARI Gene/AutismKB 2.0 |
|---|---|---|---|
| “High confidence” ASD-genes | |||
| USP7 | 16p13.2 (P3) | Ubiquitin-specific protease; regulates ubiquitination processes | 2/4 |
| GRIN2A | 16p13.2 (P3) | Subunit 2A of the glutamate N-Methyl-D-Aspartate (NMDA) receptor | 2/10 |
| RBFOX1 | 16p13.3 (P3) | RNA-binding protein that regulates alternative splicing events | 2/28 |
| DMD | Xp21.1 (P25) | Component of the dystrophin-glycoprotein complex (DGC), which bridges the inner cytoskeleton and the extracellular matrix | S/28 |
| SHOX | Xp22.33 (P29) | Belongs to the paired homeobox family, nuclear transcription factors involved in cell-cycle and growth regulation | 2/2 |
| NRXN1 | 2p16.2 (P29) | Cell adhesion molecule, form a complex with neuroligins at synapses in the central nervous system required for neurotransmission and involved in the formation of synaptic contacts. | 1/68 |
| Suggestive or “low confidence” candidate ASD-genes | |||
| TSPAN7 | Xp11.4 (P1) | Member of the tetraspanin family, encodes a cell surface glycoprotein that complex with integrins. It may have a role in neurite outgrowth and Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor trafficking | 3/2 |
| ITGB2 | 21q22.3 (P4) | Integrin B2, adhesion molecule implicated in synaptic pruning | NR/3 |
| TRPM2 | 21q22.3 (P4) | Voltage-independent cation channel, mediates sodium and calcium ion influx in response to oxidative stress; modulates oxytocin release. | NR/11 |
| ADARB1 | 21q22.3 (P4) | Protein involved in the editing of the RNA of glutamate, serotonin and Gamma-Aminobutyric Acid (GABA) receptors, and potassium voltage-gated channels. | 5/1 |
| SUMO3 | 21q22.3 (P4) | Involved in SUMOylation of proteins, a post-translational modification that modulates the activity of several neuronal transcription factors | NR/0 |
| RFX7 | 15q21.3 (P9) | Transcription factor | NR/4 |
| NEDD4 | 15q21.3 (P9) | Protein involved in the ubiquitin proteasome system. It plays a critical role in the ubiquitination and degradation of AMPA receptors, endocytic machinery components and Phosphatase and Tensin Homolog (PTEN) protein. | NR/4 |
| PCDH15 | 10q21.1 (P11) | Member of the cadherin superfamily, membrane proteins that mediate cellular adhesion | 3/16 |
| ACCN1 (ASIC2) | 17q12 (P13) | Non-voltage-dependent Na+ channel; facilitates Acid-Sensing Ion Channel (ASIC) localization to synapses interacting with synaptic scaffolding proteins as Postsynaptic Density Protein 95 (PSD95) | NR/7 |
| TMEM132E | 17q12 (P13) | Neural adhesion molecule | NR/NR |
| CADM2 | 3p12.1 (P14) | Adhesion molecule involved in synapse organization, providing regulated trans-synaptic adhesion. | 3/0 |
| GRM7 | 3p26.1 (P14) | Metabotropic glutamate receptor mGluR7 | 3/12 |
| IMMP2L | 7q31.1 (P15) | Subunit of an inner mitochondrial membrane peptidase complex involved in processing of mitochondrial proteins | 3/10 |
| PTPRD | 9p23p24 (P22) | Receptor protein tyrosine phosphatase, induces pre- and post-synaptic differentiation and regulates neurogenesis. Interacts with proteins involved in intellectual disability/ASD as IL1RAP and IL1RAPL1 and proteins of the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway. | NR/7 |
| ST6GAL2 | 2q12.3 (P27) | Encodes a sialyltransferase mostly expressed in embryonic and adult brain. CNVs were reported in autism studies. | NR/2 |
| Type of CNVs | Inheritance | ||||
|---|---|---|---|---|---|
| Duplication | Deletion | De novo | Paternal | Maternal | |
| (n = 17) | (n = 18) | (n = 8) | (n = 12) | (n = 9) | |
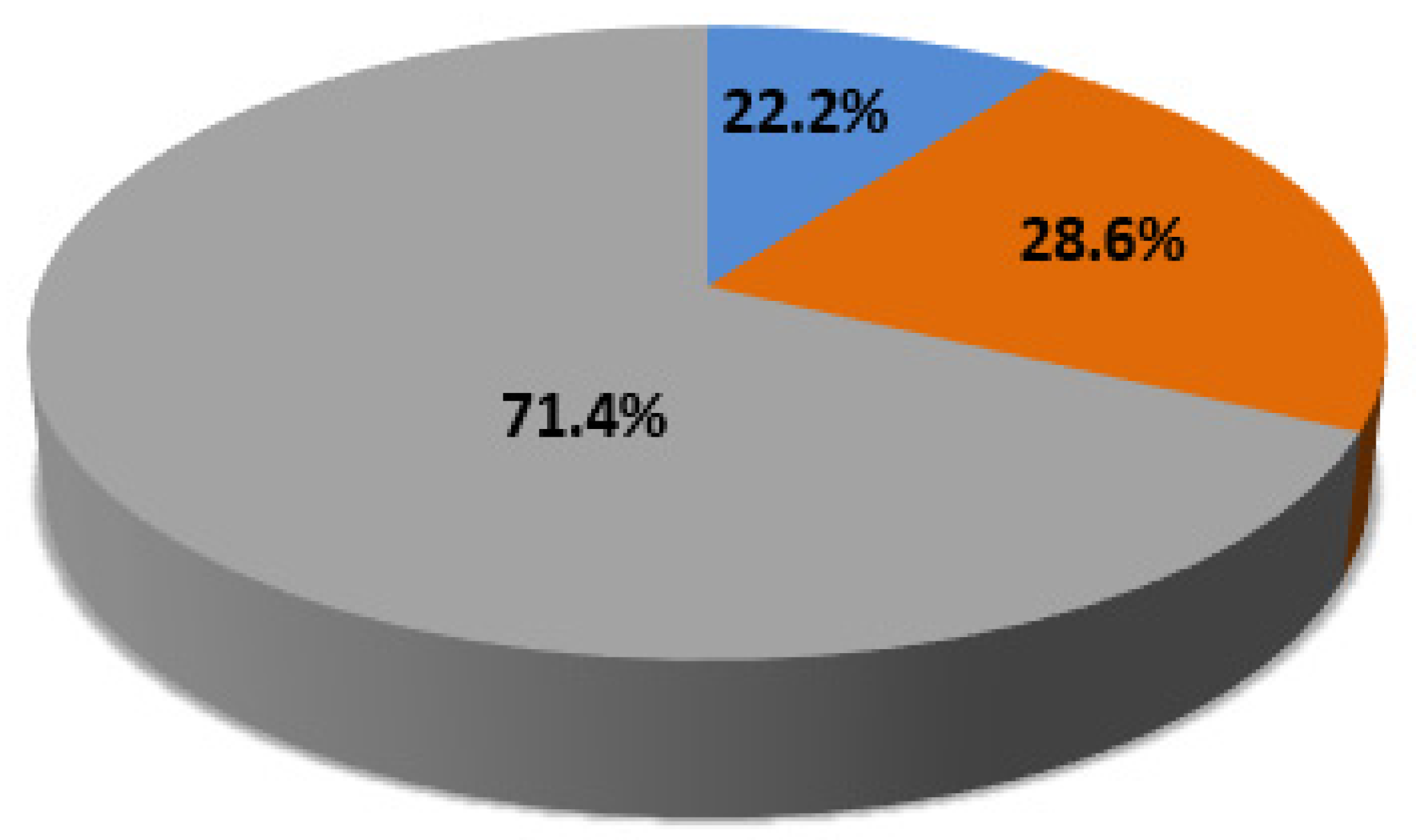
| C-CNVs | 13/25 (52%) | 12/25 (48%) | 8/21 (38.1%) | 6/21 (28.6%) | 7/21 (33.3%) |
| (n = 25) | |||||
| N-CNVs | 4/10 (40%) | 6/10 (60%) | 0/8 (0%) | 6/8 (75%) | 2/8 (25%) |
| (n = 10) | |||||
| Total | 17/35 (48.6%) | 18/35 (51.4%) | 8/29 (27.6%) | 12/29 (41.4%) | 9/29 (31%) |
| C-CNVs | N-CNVs | w-CNVs | |
|---|---|---|---|
| (n = 20) | (n = 9) | (n = 61) | |
| Mean age at the last examination in months (SD) | 66.95 (38.55) | 47.11 (15.57) | 58.16 (40.19) |
| IQ > 70 | 9/20 (45%) | 4/8 (50%) | 32/59 (54.2%) |
| IQ ≤ 70 | 11/20 (55%) | 4/8 (50%) | 27/59 (45.8%) |
| Verbal | 13/20 (65%) | 5/9 (55.6%) | 45/61 (73.7%) |
| Non-verbal | 7/20 (35%) | 4/9 (44.4%) | 16/61 (26.3%) |
| Mean ADOS-CSS: | - | - | - |
| Mean SA-CSS (SD) | 6.38 (2.26) | 7.50 (1.64) | 6.81 (2.45) |
| Mean RRB-CSS (SD) | 6.08 (2.14) | 5.50 (3.83) | 7.75 (1.92) |
| Mean Global-CSS (SD) | 5.69 (2.25) | 6.83 (2.64) | 6.77 (2.35) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderoni, S.; Ricca, I.; Balboni, G.; Cagiano, R.; Cassandrini, D.; Doccini, S.; Cosenza, A.; Tolomeo, D.; Tancredi, R.; Santorelli, F.M.; et al. Evaluation of Chromosome Microarray Analysis in a Large Cohort of Females with Autism Spectrum Disorders: A Single Center Italian Study. J. Pers. Med. 2020, 10, 160. https://doi.org/10.3390/jpm10040160
Calderoni S, Ricca I, Balboni G, Cagiano R, Cassandrini D, Doccini S, Cosenza A, Tolomeo D, Tancredi R, Santorelli FM, et al. Evaluation of Chromosome Microarray Analysis in a Large Cohort of Females with Autism Spectrum Disorders: A Single Center Italian Study. Journal of Personalized Medicine. 2020; 10(4):160. https://doi.org/10.3390/jpm10040160
Chicago/Turabian StyleCalderoni, Sara, Ivana Ricca, Giulia Balboni, Romina Cagiano, Denise Cassandrini, Stefano Doccini, Angela Cosenza, Deborah Tolomeo, Raffaella Tancredi, Filippo Maria Santorelli, and et al. 2020. "Evaluation of Chromosome Microarray Analysis in a Large Cohort of Females with Autism Spectrum Disorders: A Single Center Italian Study" Journal of Personalized Medicine 10, no. 4: 160. https://doi.org/10.3390/jpm10040160
APA StyleCalderoni, S., Ricca, I., Balboni, G., Cagiano, R., Cassandrini, D., Doccini, S., Cosenza, A., Tolomeo, D., Tancredi, R., Santorelli, F. M., & Muratori, F. (2020). Evaluation of Chromosome Microarray Analysis in a Large Cohort of Females with Autism Spectrum Disorders: A Single Center Italian Study. Journal of Personalized Medicine, 10(4), 160. https://doi.org/10.3390/jpm10040160