
Identification of Differentially Expressed Genes in Different Glioblastoma Regions and Their Association with Cancer Stem Cell Development and Temozolomide Response

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohorts
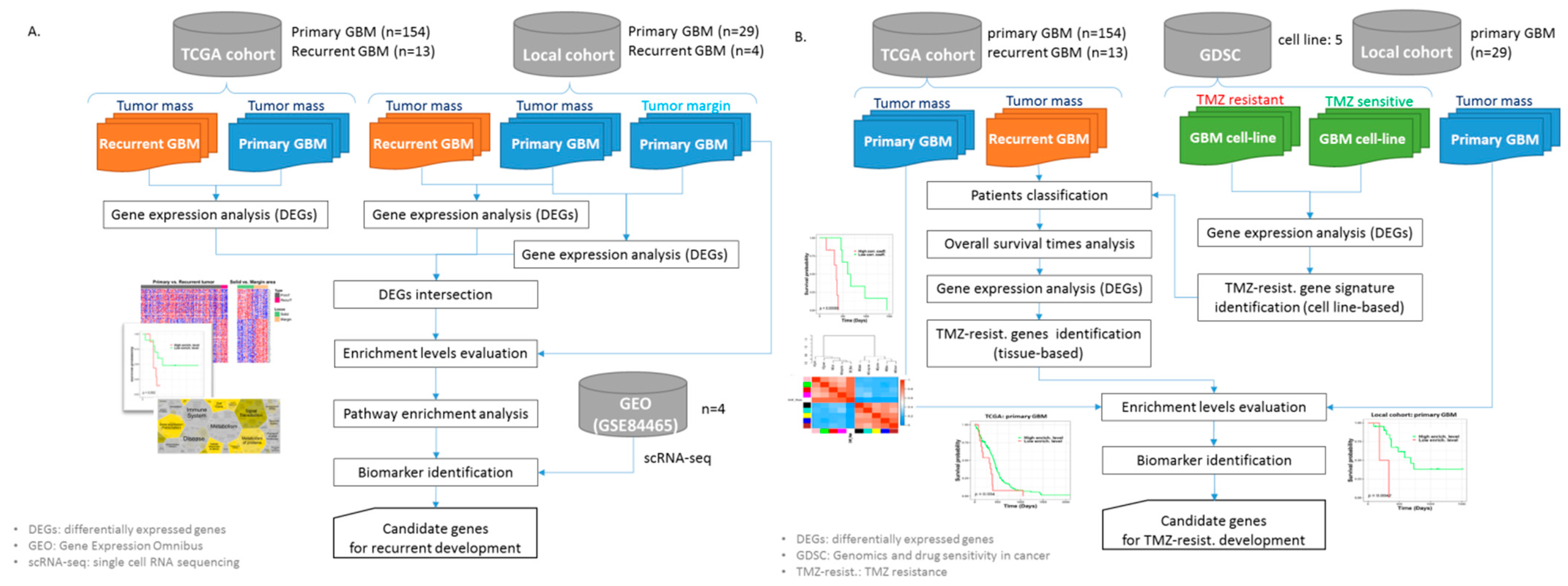
2.2. The Workflows of the Study
2.3. Genomic Data Preprocessing
2.4. Differentially Expressed Gene Analysis
2.5. Gene Set Enrichment Analysis
2.6. Identification of GBM Recurrence-Associated Gene Signatures
2.7. Identification of Tissue-Based Gene Signatures of GBM Sample Responses to TMZ
2.7.1. Cell Line-Based TMZ-Resistant Gene Signatures
2.7.2. Tumor Tissue-Based TMZ-Resistant Gene Signatures
3. Results
3.1. Patient Characteristics
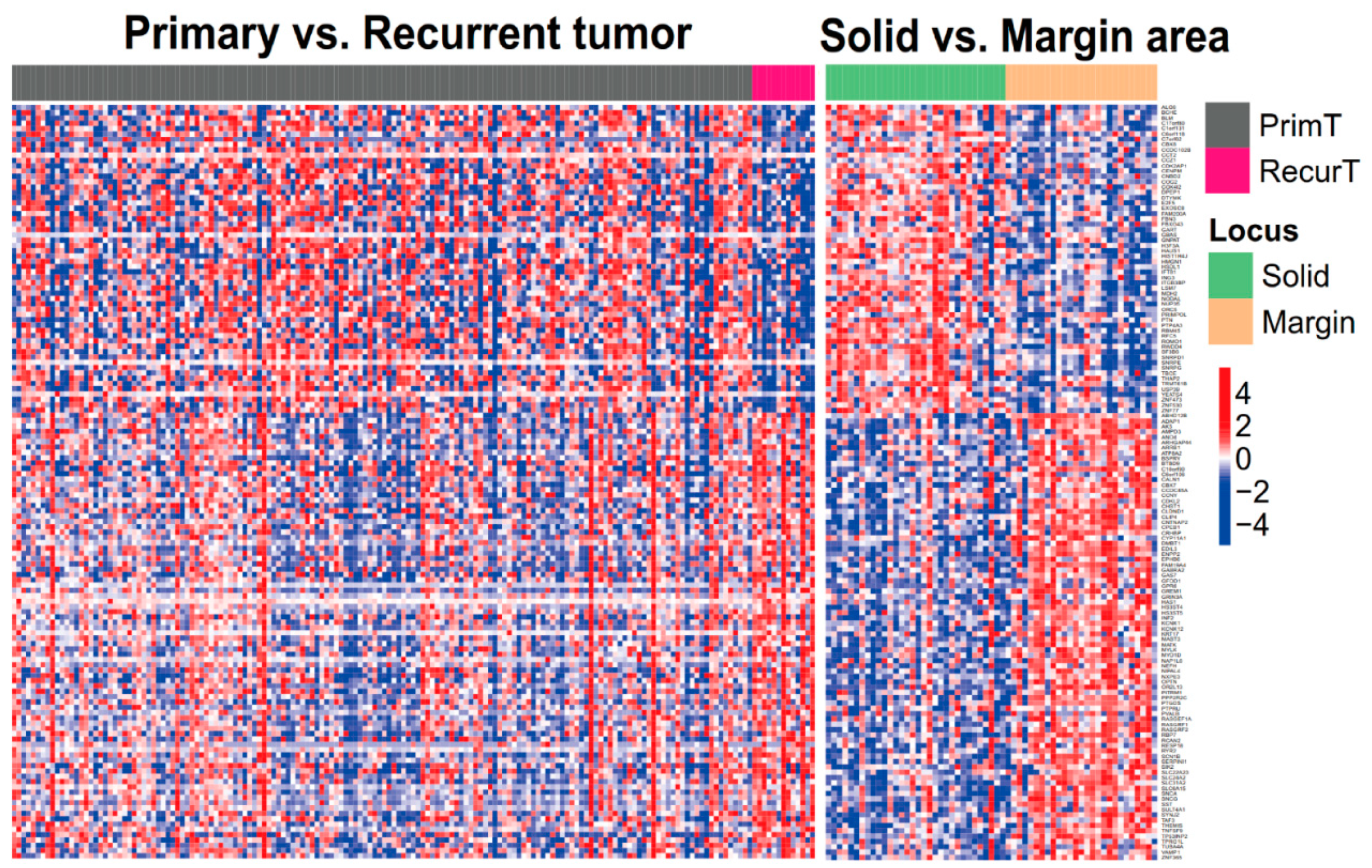
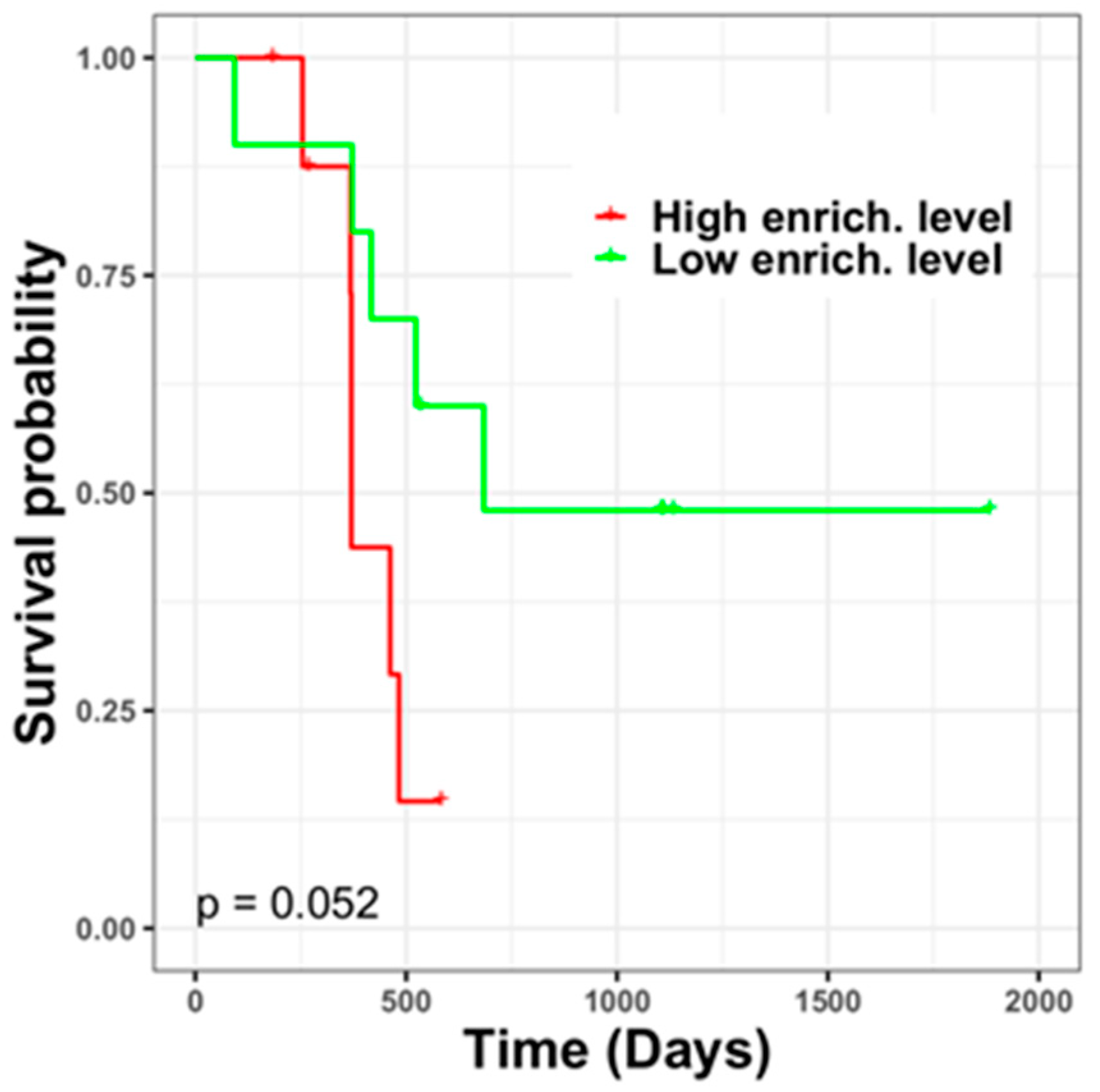
3.2. GBM Recurrence-Associated Gene Signatures in Tumor Margins
3.3. Pathway Involvement of GBM Recurrence-Associated Gene Signatures and Potential Biomarkers of Their Regulatory Network
3.4. GBM Cell Line-Based TMZ Resistance-Associated Gene Signatures
3.5. GBM Tissue-Based TMZ Resistance-Associated Gene Signatures
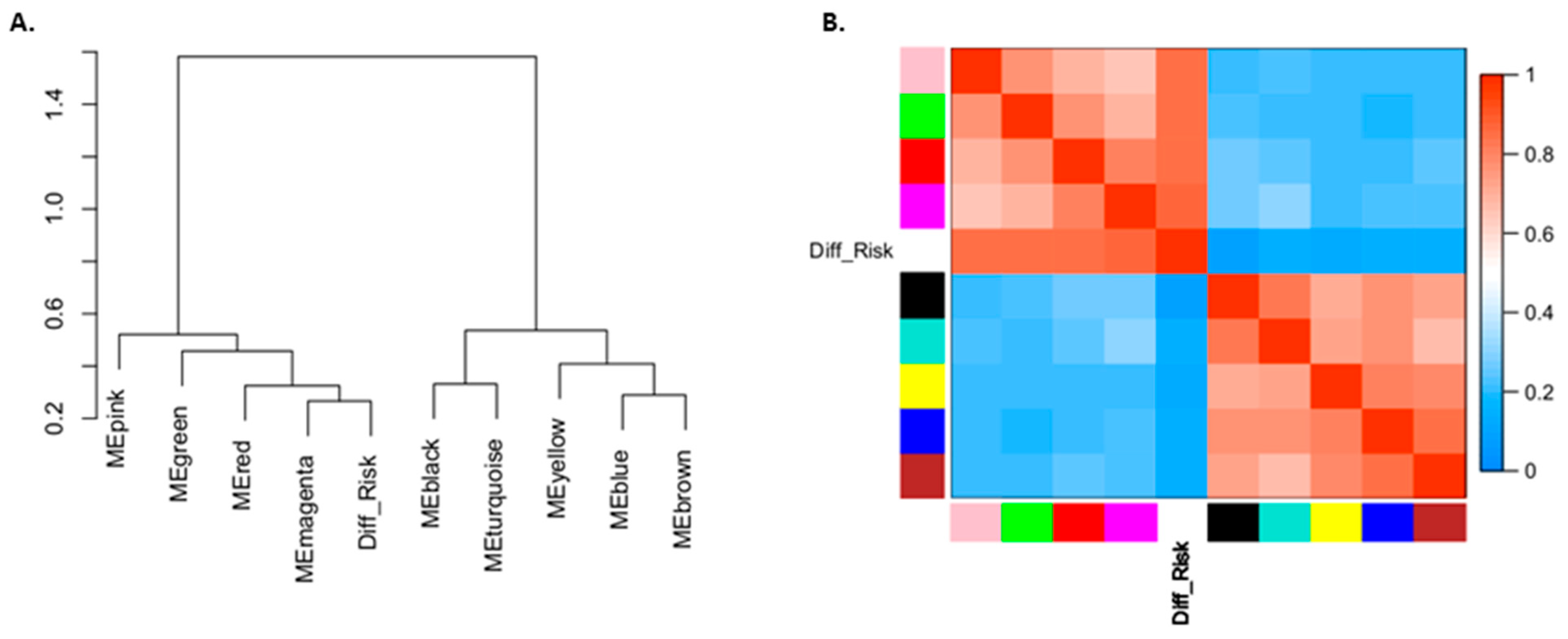
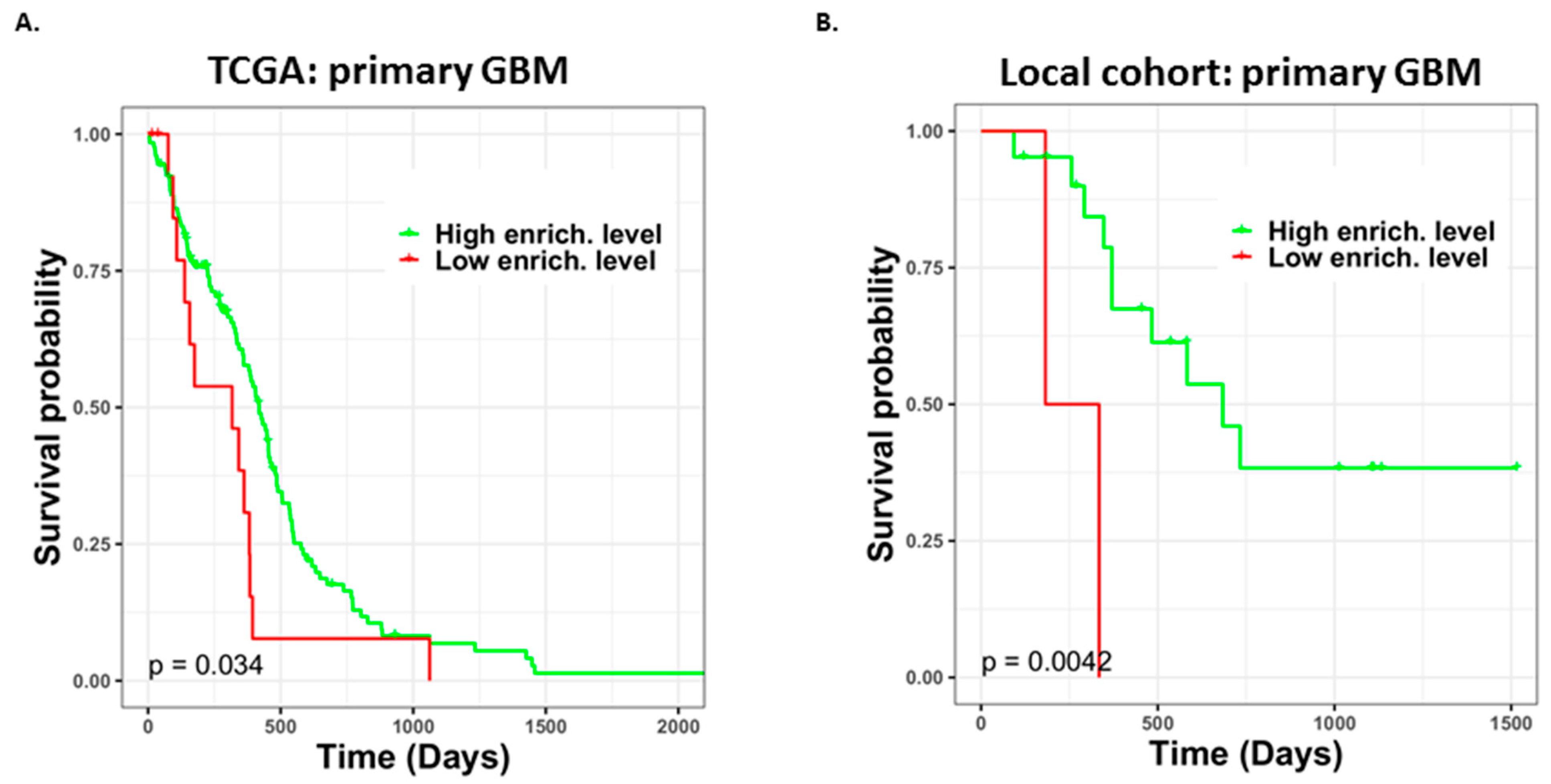
3.6. Different TMZ Resistance Risks and Potentially Related Biomarkers within the Tumor Mass of Primary GBM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15 (Suppl. 2), ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, V.; Rabiee, A.; Khademi, F. Glioblastoma multiforme: A glance at advanced therapies based on nanotechnology. J. Chemother. 2020, 32, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, E.; Yamamoto, T.; Matsumura, A. Prospect of Immunotherapy for Glioblastoma: Tumor Vaccine, Immune Checkpoint Inhibitors and Combination Therapy. Neurol. Med. Chir. 2017, 57, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 133, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Draaisma, K.; Chatzipli, A.; Taphoorn, M.; Kerkhof, M.; Weyerbrock, A.; Sanson, M.; Hoeben, A.; Lukacova, S.; Lombardi, G.; Leenstra, S.; et al. Molecular Evolution of IDH Wild-Type Glioblastomas Treated With Standard of Care Affects Survival and Design of Precision Medicine Trials: A Report From the EORTC 1542 Study. J. Clin. Oncol. 2020, 38, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Tabatabai, G.; Kastner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.; Kroon, P.; Zhang, Y.; Galan-Ganga, M.; Bao, Z.; Wang, Z.; Liu, H.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 3883. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.M.; Kang, S.H.; Park, C.K.; Jung, S.; Park, E.S.; Lee, J.S.; Kim, S.H.; Woo, H.G. Recurrent Glioblastomas Reveal Molecular Subtypes Associated with Mechanistic Implications of Drug-Resistance. PLoS ONE 2015, 10, e0140528. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef] [PubMed]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the mechanisms of Temozolomide resistance in gliomas and the strategies to overcome the resistance. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188616. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Lenz, G.; Wink, M.R. EMT-like process in glioblastomas and reactive astrocytes. Neurochem. Int. 2019, 122, 139–143. [Google Scholar] [CrossRef]
- Chen, L.; Guo, P.; He, Y.; Chen, Z.; Chen, L.; Luo, Y.; Qi, L.; Liu, Y.; Wu, Q.; Cui, Y.; et al. HCC-derived exosomes elicit HCC progression and recurrence by epithelial-mesenchymal transition through MAPK/ERK signalling pathway. Cell Death Dis. 2018, 9, 513. [Google Scholar] [CrossRef] [Green Version]
- Hide, T.; Komohara, Y.; Miyasato, Y.; Nakamura, H.; Makino, K.; Takeya, M.; Kuratsu, J.I.; Mukasa, A.; Yano, S. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine 2018, 30, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Bastola, S.; Pavlyukov, M.S.; Yamashita, D.; Ghosh, S.; Cho, H.; Kagaya, N.; Zhang, Z.; Minata, M.; Lee, Y.; Sadahiro, H.; et al. Glioma-initiating cells at tumor edge gain signals from tumor core cells to promote their malignancy. Nat. Commun. 2020, 11, 4660. [Google Scholar] [CrossRef]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- El-Khayat, S.M.; Arafat, W.O. Therapeutic strategies of recurrent glioblastoma and its molecular pathways ‘Lock up the beast’. Ecancermedicalscience 2021, 15, 1176. [Google Scholar] [CrossRef]
- Rabe, M.; Dumont, S.; Alvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Sery, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis 2020, 11, 19. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 2019, 11, 921. [Google Scholar] [CrossRef] [Green Version]
- Dubey, P.K.; Masuda, K.; Nyati, K.K.; Uz Zaman, M.M.; Chalise, J.P.; Millrine, D.; Kai, W.; Ripley, B.; Kishimoto, T. Arid5a-deficient mice are highly resistant to bleomycin-induced lung injury. Int. Immunol. 2017, 29, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Eduardo da Silva, L.; Russo, L.C.; Forti, F.L. Overactivated Cdc42 acts through Cdc42EP3/Borg2 and NCK to trigger DNA damage response signaling and sensitize cells to DNA-damaging agents. Exp. Cell Res. 2020, 395, 112206. [Google Scholar] [CrossRef]
- Hu, K.; Li, J.; Wu, G.; Zhou, L.; Wang, X.; Yan, Y.; Xu, Z. The novel roles of virus infection-associated gene CDKN1A in chemoresistance and immune infiltration of glioblastoma. Aging 2021, 13, 6662–6680. [Google Scholar] [CrossRef]
- Bandey, I.; Chiou, S.H.; Huang, A.P.; Tsai, J.C.; Tu, P.H. Progranulin promotes Temozolomide resistance of glioblastoma by orchestrating DNA repair and tumor stemness. Oncogene 2015, 34, 1853–1864. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Cohort | TCGA Cohort | Local Cohort | GSE84465 | |||
|---|---|---|---|---|---|---|
| Tumor Type | Primary (n = 154) | Recurrent (n = 13) | Primary (n = 29) | Recurrent (n = 4) | Primary (n = 4) | |
| IDH | WT | 139 | 12 | 21 | 0 | 3 |
| Mutant | 10 | 1 | 0 | 0 | 0 | |
| n/a | 5 | 0 | 8 | 4 | 1 | |
| Age | Median (Q1–Q3) | 60.0 (52.0–70.0) | 58.0 (48.0–63.0) | 62.0 (54.0–70.0) | 53.5 (45.8–59.0) | n/a |
| Gender | Male | 99 | 8 | 19 | 2 | 0 |
| Female | 54 | 5 | 10 | 2 | 0 | |
| n/a | 1 | 0 | 0 | 0 | 4 | |
| Survival (Days) | Median (Q1–Q3) | 342.5 (153.0–515.5) | 399.0 (317.0–603.0) | 413.0 (286.2–696.2) | 396.5 (294.2–624.2) | n/a |
| Tumor Margin Relative to Tumor Mass (Diff. Regions) | High Risk Relative to Low Risk (in the Tumor Margin) | |||
|---|---|---|---|---|
| Log2FC | p | Log2FC | p | |
| DVL1 | 0.199 | 0.048 | 1.147 | 0.000 |
| PRKACB | 0.930 | 0.000 | 1.434 | 0.000 |
| ARRB1 | 1.145 | 0.001 | 1.155 | 0.017 |
| APC | 0.711 | 0.003 | 1.046 | 0.006 |
| MAPK9 | 0.495 | 0.001 | 0.863 | 0.008 |
| CAMK2A | 2.875 | 0.042 | 5.115 | 0.008 |
| PRKCB | 2.710 | 0.001 | 2.749 | 0.002 |
| CACNA1A | 1.074 | 0.030 | 1.811 | 0.000 |
| ERBB4 | 1.181 | 0.005 | 0.875 | 0.017 |
| RASGRF1 | 1.694 | 0.000 | 1.915 | 0.000 |
| NF1 | 0.132 | 0.045 | 0.690 | 0.003 |
| RPS6KA2 | 1.123 | 0.000 | 0.744 | 0.009 |
| MAPK8IP2 | 0.746 | 0.007 | 1.533 | 0.004 |
| PPM1A | 0.464 | 0.005 | 0.741 | 0.000 |
| Astrocyte | Immune Cell | Neoplastic | Neuron | Oligodendrocyte | OPC | Vascular | Total Expr. Cells | |
|---|---|---|---|---|---|---|---|---|
| DVL1 | 9 | 21 | 7 | 7 | 7 | 44 | n/a | 95 |
| ARRB1 | 20 | 61 | 2 | 4 | 1 | 7 | n/a | 95 |
| PRKACB | 35 | 76 | 29 | 19 | 31 | 280 | 1 | 471 |
| APC | 76 | 66 | 34 | 17 | 19 | 199 | n/a | 411 |
| MAPK9 | 14 | 60 | 12 | 11 | 7 | 71 | 1 | 176 |
| CAMK2A | 3 | 1 | n/a | 14 | n/a | 21 | n/a | 39 |
| PRKCB | n/a | 109 | 1 | 15 | 4 | 96 | n/a | 225 |
| CACNA1A | 2 | 153 | 3 | 7 | 1 | 161 | n/a | 327 |
| ERBB4 | 44 | 1 | 15 | 19 | 8 | 166 | n/a | 253 |
| RASGRF1 | n/a | 6 | 2 | 16 | 2 | 4 | n/a | 30 |
| NF1 | 48 | 148 | 19 | 15 | 15 | 181 | 2 | 428 |
| RPS6KA2 | 27 | 92 | 3 | 17 | 11 | 84 | 1 | 235 |
| MAPK8IP2 | 11 | 2 | 2 | 9 | n/a | 61 | n/a | 85 |
| PPM1A | 16 | 52 | 11 | 8 | 9 | 110 | 1 | 207 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, J.B.-K.; Lee, T.-Y.; Cheng, S.-J.; Lee, G.A.; Chen, Y.-C.; Le, N.Q.K.; Huang, S.-W.; Kuo, D.-P.; Li, Y.-T.; Chang, T.-H.; et al. Identification of Differentially Expressed Genes in Different Glioblastoma Regions and Their Association with Cancer Stem Cell Development and Temozolomide Response. J. Pers. Med. 2021, 11, 1047. https://doi.org/10.3390/jpm11111047
Hsu JB-K, Lee T-Y, Cheng S-J, Lee GA, Chen Y-C, Le NQK, Huang S-W, Kuo D-P, Li Y-T, Chang T-H, et al. Identification of Differentially Expressed Genes in Different Glioblastoma Regions and Their Association with Cancer Stem Cell Development and Temozolomide Response. Journal of Personalized Medicine. 2021; 11(11):1047. https://doi.org/10.3390/jpm11111047
Chicago/Turabian StyleHsu, Justin Bo-Kai, Tzong-Yi Lee, Sho-Jen Cheng, Gilbert Aaron Lee, Yung-Chieh Chen, Nguyen Quoc Khanh Le, Shiu-Wen Huang, Duen-Pang Kuo, Yi-Tien Li, Tzu-Hao Chang, and et al. 2021. "Identification of Differentially Expressed Genes in Different Glioblastoma Regions and Their Association with Cancer Stem Cell Development and Temozolomide Response" Journal of Personalized Medicine 11, no. 11: 1047. https://doi.org/10.3390/jpm11111047
APA StyleHsu, J. B.-K., Lee, T.-Y., Cheng, S.-J., Lee, G. A., Chen, Y.-C., Le, N. Q. K., Huang, S.-W., Kuo, D.-P., Li, Y.-T., Chang, T.-H., & Chen, C.-Y. (2021). Identification of Differentially Expressed Genes in Different Glioblastoma Regions and Their Association with Cancer Stem Cell Development and Temozolomide Response. Journal of Personalized Medicine, 11(11), 1047. https://doi.org/10.3390/jpm11111047