Gut Microbiota Composition across Normal Range Prostate-Specific Antigen Levels

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and Group Definitions
2.2. Sample Collection, DNA Extraction, and 16S rRNA Gene Sequencing
2.3. Microbial Profiling and Statistical Analysis
3. Results
3.1. Subject Demographics
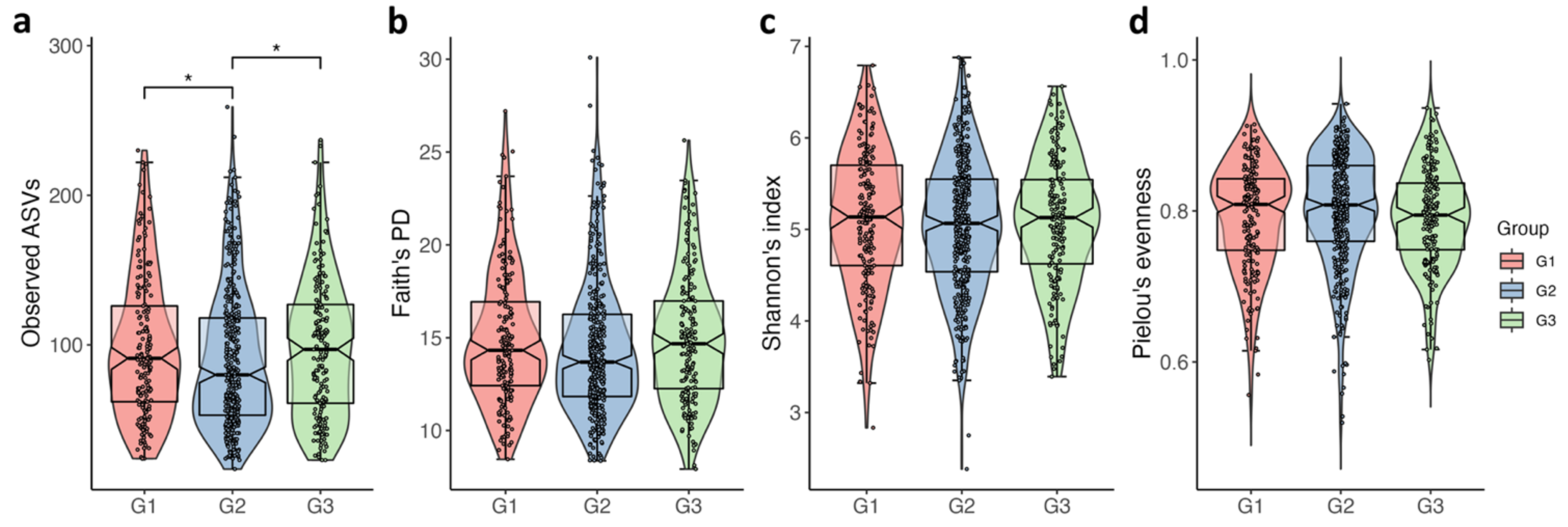
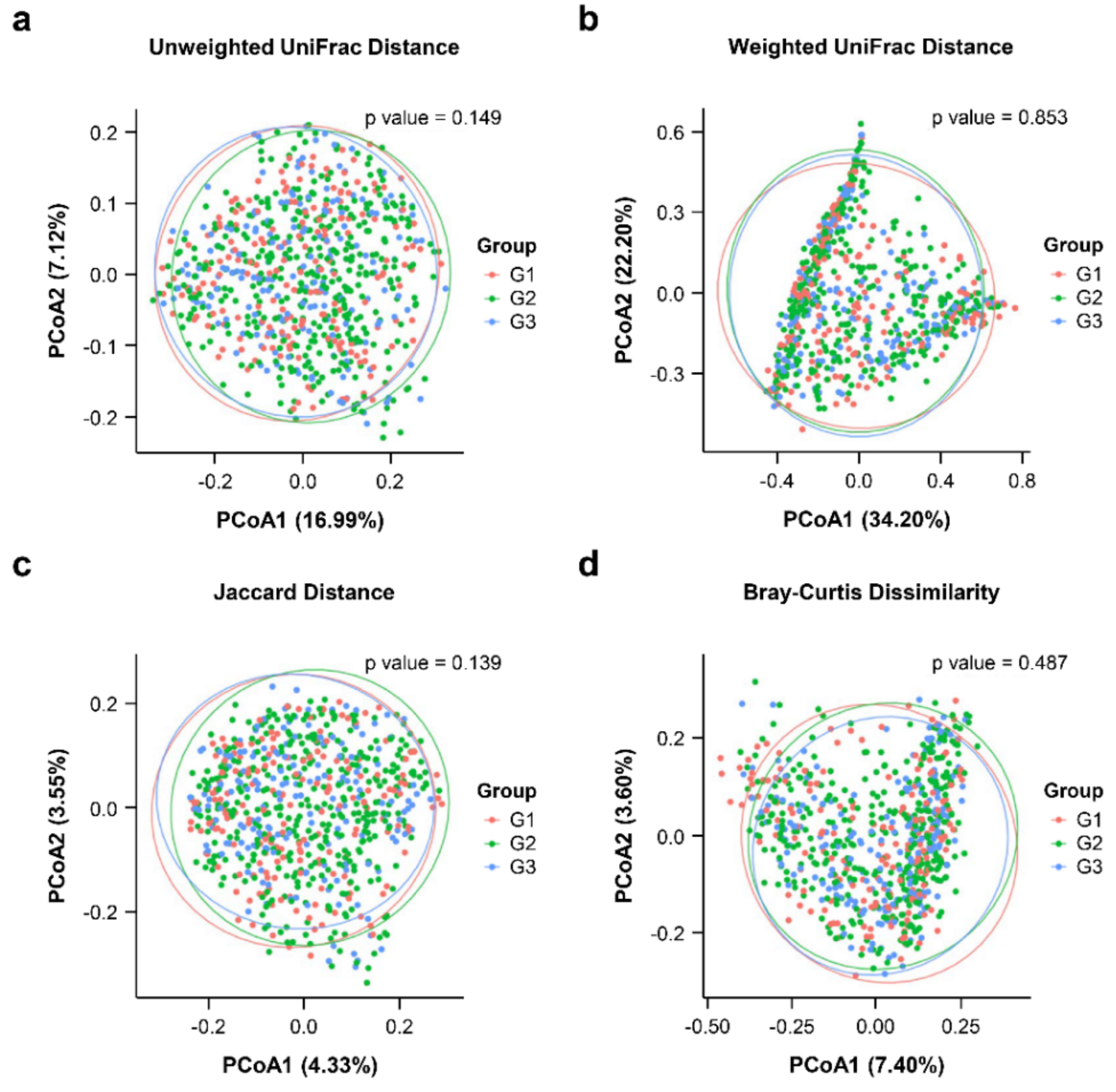
3.2. Comparison Microbial Diversity of among the PSA Groups
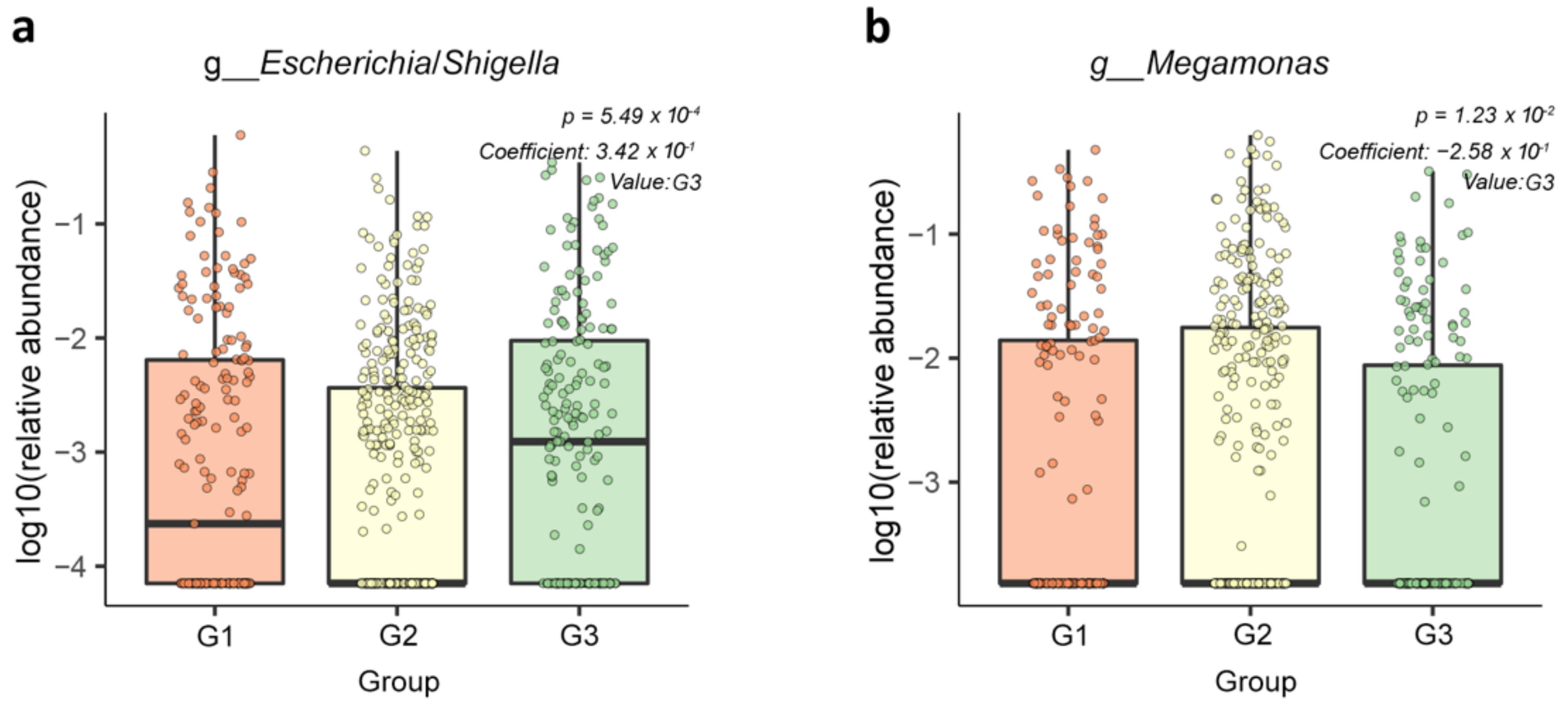
3.3. Fecal Bacterial Community Abundance among the PSA Groups
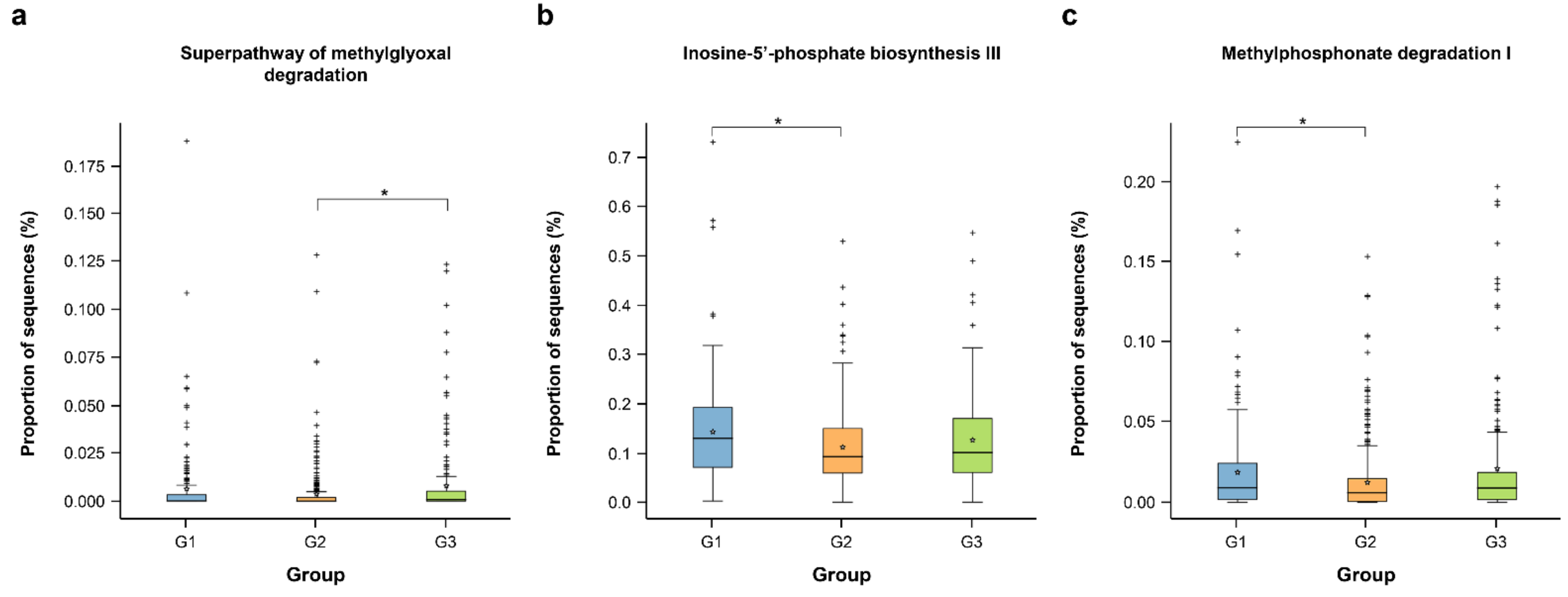
3.4. Predicted Functional Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Coetzee, G.A. Prostate specific antigen gene regulation by androgen receptor. J. Cell Biochem. 2004, 93, 233–241. [Google Scholar] [CrossRef]
- Wang, L.G.; Liu, X.M.; Kreis, W.; Budman, D.R. Down-regulation of prostate-specific antigen expression by finasteride through inhibition of complex formation between androgen receptor and steroid receptor-binding consensus in the promoter of the PSA gene in LNCaP cells. Cancer Res. 1997, 57, 714–719. [Google Scholar]
- Chang, Y.; Kim, J.H.; Noh, J.W.; Cho, Y.S.; Park, H.J.; Joo, K.J.; Ryu, S. Prostate-specific antigen within the reference range, subclinical coronary atherosclerosis, and cardiovascular mortality. Circ. Res. 2019, 124, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Noh, J.W.; Chang, Y.; Lee, H.Y.; Park, J.J.; Ryu, S.; Kim, J.H. Association between prostate-specific antigen and serum testosterone: A systematic review and meta-analysis. Andrology 2020, 8, 1194–1213. [Google Scholar] [CrossRef]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Poutahidis, T.; Springer, A.; Levkovich, T.; Qi, P.; Varian, B.J.; Lakritz, J.R.; Ibrahim, Y.M.; Chatzigiagkos, A.; Alm, E.J.; Erdman, S.E. Probiotic microbes sustain youthful serum testosterone levels and testicular size in aging mice. PLoS ONE 2014, 9, e84877. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Park, Y.H.; Sim, M.; Kim, S.A.; Joung, H.; Shin, D.M. Serum level of sex steroid hormone is associated with diversity and profiles of human gut microbiome. Res. Microbiol. 2019, 170, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprient 2018, 2, e27295. [Google Scholar] [CrossRef]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinform. Online 2007, 2, 121–128. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vazquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Cross, T.L.; Kasahara, K.; Rey, F.E. Sexual dimorphism of cardiometabolic dysfunction: Gut microbiome in the play? Mol. Metab. 2018, 15, 70–81. [Google Scholar] [CrossRef]
- Collden, H.; Landin, A.; Wallenius, V.; Elebring, E.; Fandriks, L.; Nilsson, M.E.; Ryberg, H.; Poutanen, M.; Sjogren, K.; Vandenput, L.; et al. The gut microbiota is a major regulator of androgen metabolism in intestinal contents. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E1182–E1192. [Google Scholar] [CrossRef]
- Moreno-Indias, I.; Sánchez-Alcoholado, L.; Sánchez-Garrido, M.; Martín-Núñez, G.M.; Pérez-Jiménez, F.; Tena-Sempere, M.; Tinahones, F.J.; Queipo-Ortuño, M.I. Neonatal androgen exposure causes persistent gut microbiota dysbiosis related to metabolic disease in adult female rats. Endocrinology 2016, 157, 4888–4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, S.B.; Sarsour, N.; Salehi, M.; Schroering, A.; Mell, B.; Joe, B.; Hill, J.W. Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes 2018, 9, 400–421. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Hanaoka, R.; Horiuchi, H.; Kitakaze, T.; Mitani, T.; Inui, H.; Yamaji, R. Castration influences intestinal microflora and induces abdominal obesity in high-fat diet-fed mice. Sci. Rep. 2016, 6, 23001. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Elkins, C.A.; Mullis, L.B. Mammalian steroid hormones are substrates for the major RND- and MFS-type tripartite multidrug efflux pumps of Escherichia coli. J. Bacteriol. 2006, 188, 1191–1195. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Gao, J.; Wu, J.; Zhou, Y.; Fu, H.; Ke, S.; Yang, H.; Chen, C.; Huang, L. Host gender and androgen levels regulate gut bacterial taxa in pigs leading to sex-biased serum metabolite profiles. Front Microbiol. 2019, 10, 1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, V.; Furuya, E.; Nishikaze, O. Hydrolysis of steroid glucuronides with beta-glucuronidase preparations from bovine liver, Helix pomatia, and E. coli. Clin. Chem. 1977, 23, 532–535. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Burrows, M.; Khan, A.A.; Graham, L.; Volchkov, P.; Becker, L.; Antonopoulos, D.; Umesaki, Y.; Chervonsky, A.V. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013, 39, 400–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Manning, S.D. Impact of age and sex on th.he composition and abundance of the intestinal microbiota in individuals with and without enteric infections. Ann. Epidemiol. 2016, 26, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy Japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Wetzels, S.; Wouters, K.; Schalkwijk, C.G.; Vanmierlo, T.; Hendriks, J.J. Methylglyoxal-derived advanced glycation endproducts in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandaraki, E.; Chatzigeorgiou, A.; Piperi, C.; Palioura, E.; Palimeri, S.; Korkolopoulou, P.; Koutsilieris, M.; Papavassiliou, A.G. Reduced ovarian glyoxalase-I activity by dietary glycotoxins and androgen excess: A causative link to polycystic ovarian syndrome. Mol. Med. 2012, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Milanesa, D.M.; Choudhury, M.S.; Mallouh, C.; Tazaki, H.; Konno, S. Methylglyoxal-induced apoptosis in human prostate carcinoma: Potential modality for prostate cancer treatment. Eur. Urol. 2000, 37, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Kuk, J.L.; Katzmarzyk, P.T.; Nichaman, M.Z.; Church, T.S.; Blair, S.N.; Ross, R. Visceral fat is an independent predictor of all-cause mortality in men. Obesity 2006, 14, 336–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Overall | Groups by PAS, ng/mL (Minimum–Maximum) | p for Trend | ||
|---|---|---|---|---|---|
| G1 (<25 Percentile) (0.13–0.58) | G2 (Interquartile) (0.58–1.16) | G3 (>75 Percentile) (1.17–3.76) | |||
| Number (male) | 759 (100) | 189 (100) | 379 (100) | 191 (100) | |
| Age (year) | 41.77 (±8.95) | 41.31 (±8.92) | 40.67 (±8.15) | 44.38 (±9.95) | <0.001 |
| BMI (kg/m2) | 24.65 (±2.79) | 25.06 (±3.03) | 24.72 (±2.78) | 24.10 (±2.44) | <0.001 |
| Waist circumference (cm) | 86.24 (±7.46) | 86.91 (±8.02) | 86.29 (±7.30) | 85.14 (±7.11) | 0.021 |
| Systolic BP (mmHg) | 114.10 (±12.29) | 115.02 (±13.67) | 114.43 (±11.68) | 112.53 (±11.94) | 0.048 |
| Diastolic BP (mmHg) | 74.37 (±9.41) | 74.58 (±10.19) | 74.39 (±9.09) | 74.10 (±9.27) | 0.625 |
| Current smoker | 213 (28.06) | 55 (29.10) | 110 (29.02) | 48 (25.13) | 0.441 |
| Alcohol intake 1 | 265 (34.91) | 67 (35.45) | 122 (32.19) | 76 (39.79) | 0.257 |
| PSA (ng/mL) | 0.97 (±0.58) | 0.44 (±0.10) | 0.83 (±0.15) | 1.77 (±0.58) | <0.001 |
| Glucose (mg/dL) | 99.72 (±20.20) | 102.24 (±25.65) | 97.97 (±16.45) | 100.70 (±20.63) | 0.463 |
| Total cholesterol (mg/dL) | 200.30 (±34.65) | 201.47 (±35.51) | 200.00 (±34.99) | 199.76 (±33.23) | 0.632 |
| LDL-C(mg/dL) | 123.75 (±30.51) | 126.20 (±30.86) | 123.03 (±30.25) | 122.68 (±30.69) | 0.257 |
| HDL-C (mg/dL) | 52.88 (±12.82) | 50.16 (±10.78) | 53.60 (±13.54) | 54.15 (±12.86) | 0.002 |
| Triglycerides (mg/dL) | 118.00 (±78.00) | 125.00 (±77.00) | 117.00 (±78.00) | 116.00 (±75.50) | 0.030 |
| hsCRP (mg/dL) | 0.73 (±1.63) | 0.65 (±1.02) | 0.69 (±1.53) | 0.87 (±2.20) | 0.178 |
| Hypertension | 142 (18.71) | 35 (18.52) | 65 (17.15) | 42 (21.99) | 0.384 |
| Diabetes | 75 (9.88) | 21 (11.11) | 32 (8.44) | 22 (11.52) | 0.890 |
| Obesity | 551 (27.40) | 58 (30.69) | 103 (27.18) | 47 (24.61) | 0.195 |
| Metabolic syndrome | 134 (17.65) | 39 (10.63) | 64 (16.89) | 31 (16.23) | 0.262 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-N.; Kim, J.-H.; Chang, Y.; Yang, D.; Kim, H.-L.; Ryu, S. Gut Microbiota Composition across Normal Range Prostate-Specific Antigen Levels. J. Pers. Med. 2021, 11, 1381. https://doi.org/10.3390/jpm11121381
Kim H-N, Kim J-H, Chang Y, Yang D, Kim H-L, Ryu S. Gut Microbiota Composition across Normal Range Prostate-Specific Antigen Levels. Journal of Personalized Medicine. 2021; 11(12):1381. https://doi.org/10.3390/jpm11121381
Chicago/Turabian StyleKim, Han-Na, Jae-Heon Kim, Yoosoo Chang, Dongmin Yang, Hyung-Lae Kim, and Seungho Ryu. 2021. "Gut Microbiota Composition across Normal Range Prostate-Specific Antigen Levels" Journal of Personalized Medicine 11, no. 12: 1381. https://doi.org/10.3390/jpm11121381