
Drug Response Diversity: A Hidden Bacterium?

Abstract
:1. Introduction
2. Impact of Gut Microbiota on Drug Effect
2.1. The Gut Microbiota Responsible for Drug Response
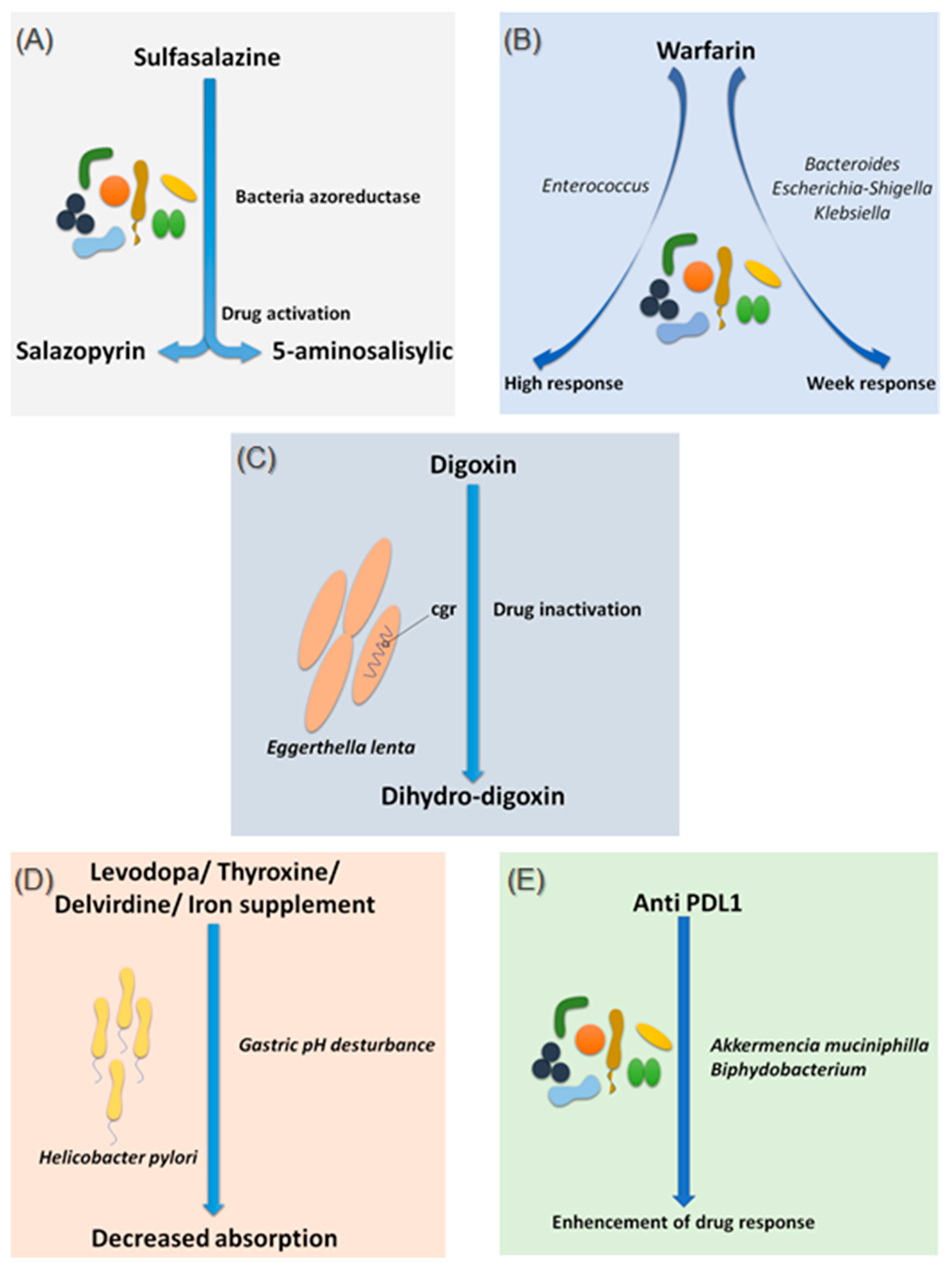
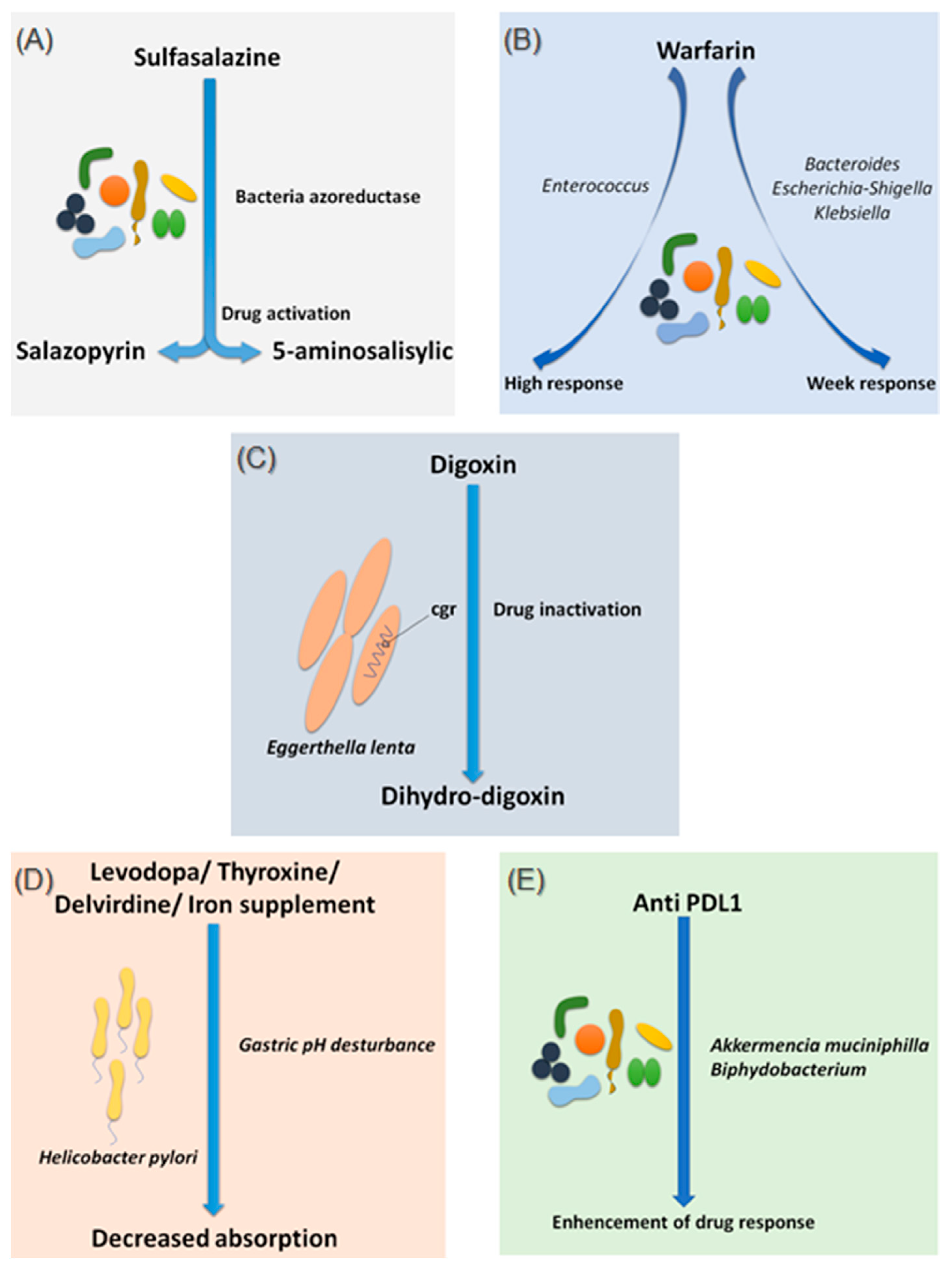
2.1.1. Sulfasalazine
2.1.2. Warfarin
2.1.3. Digoxin
2.1.4. Levodopa
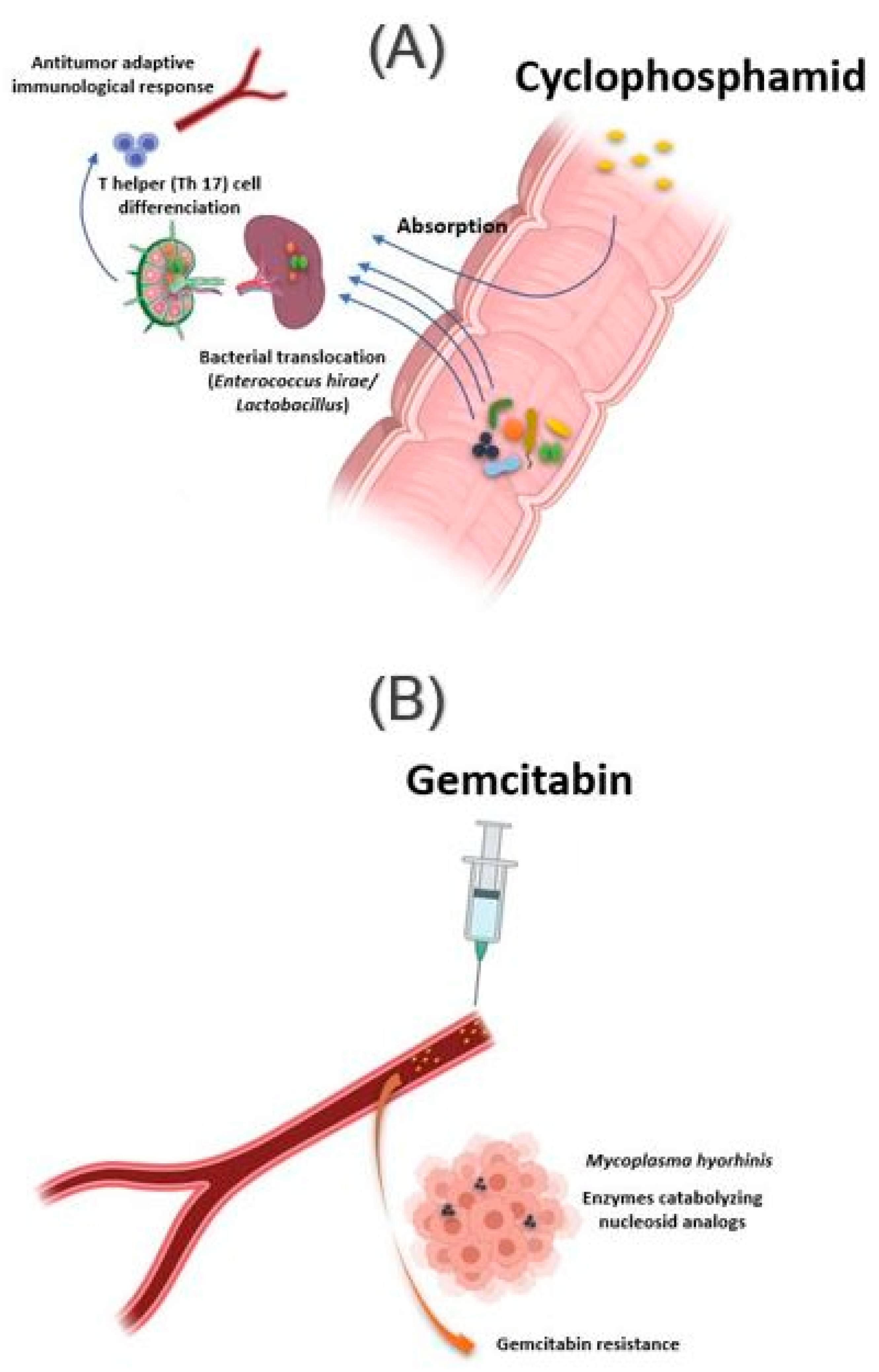
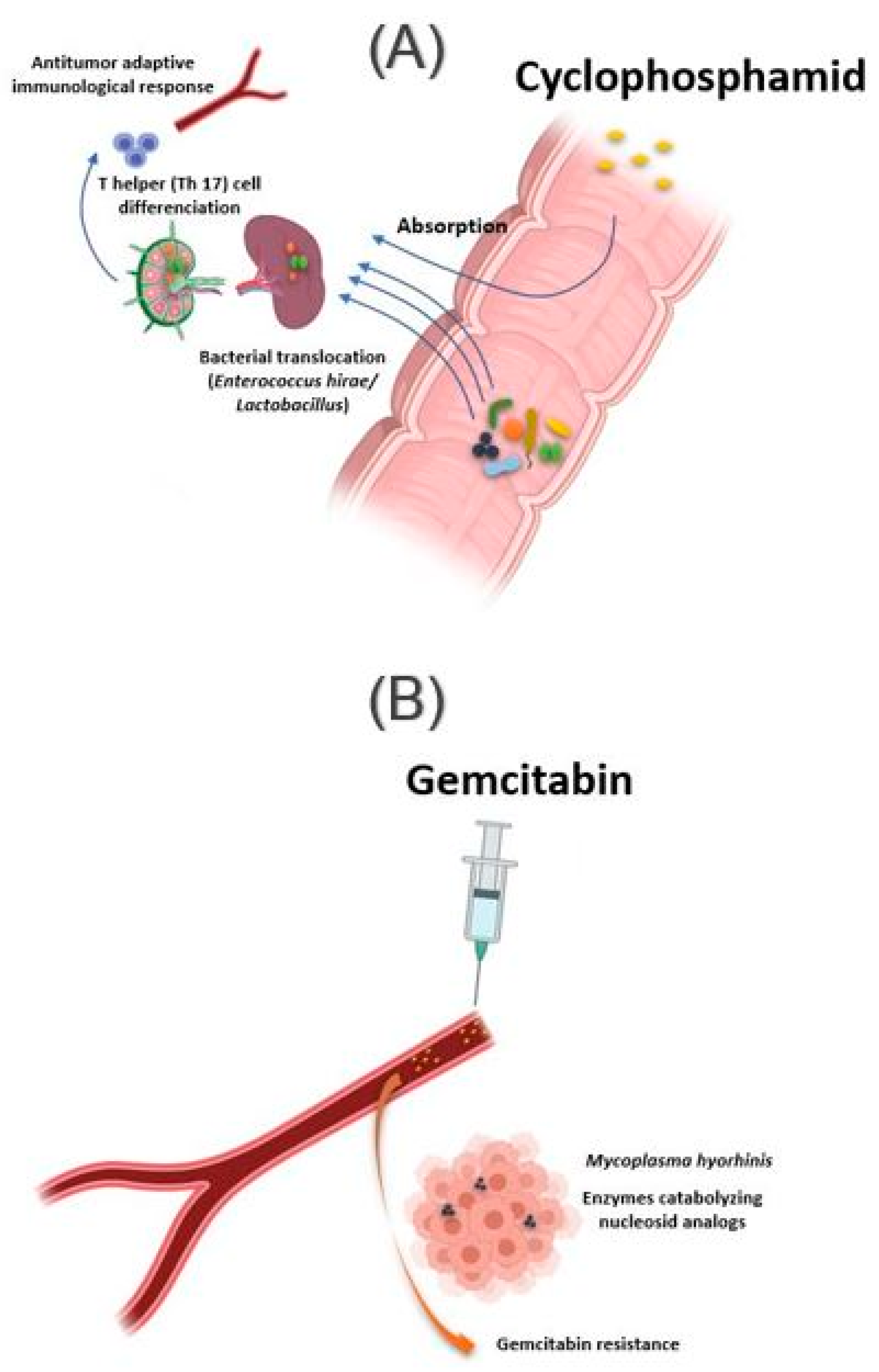
2.1.5. Chemotherapy and Immunomodulator Drugs
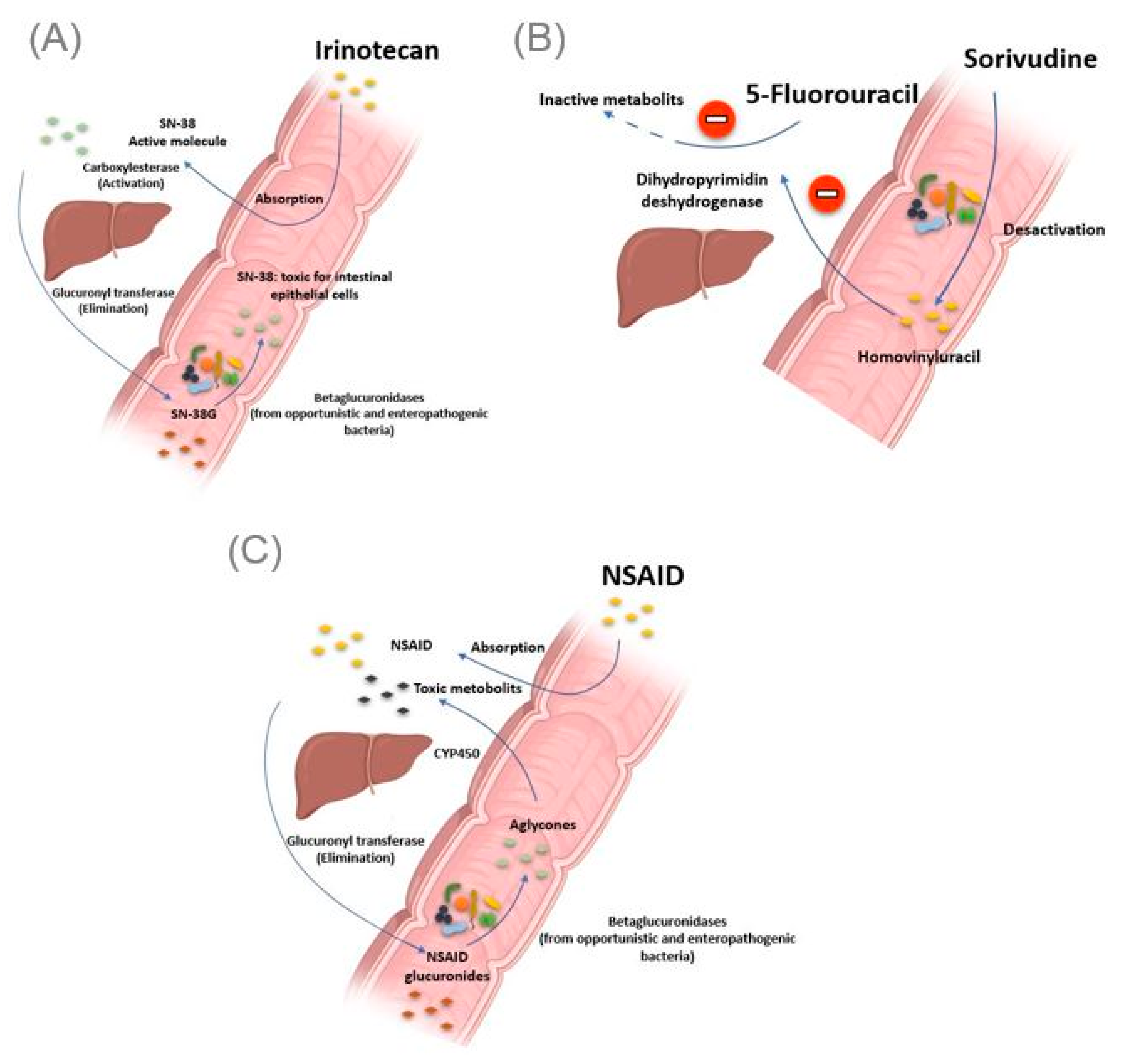
2.2. The Gut Microbiota Causing Drug Toxicity
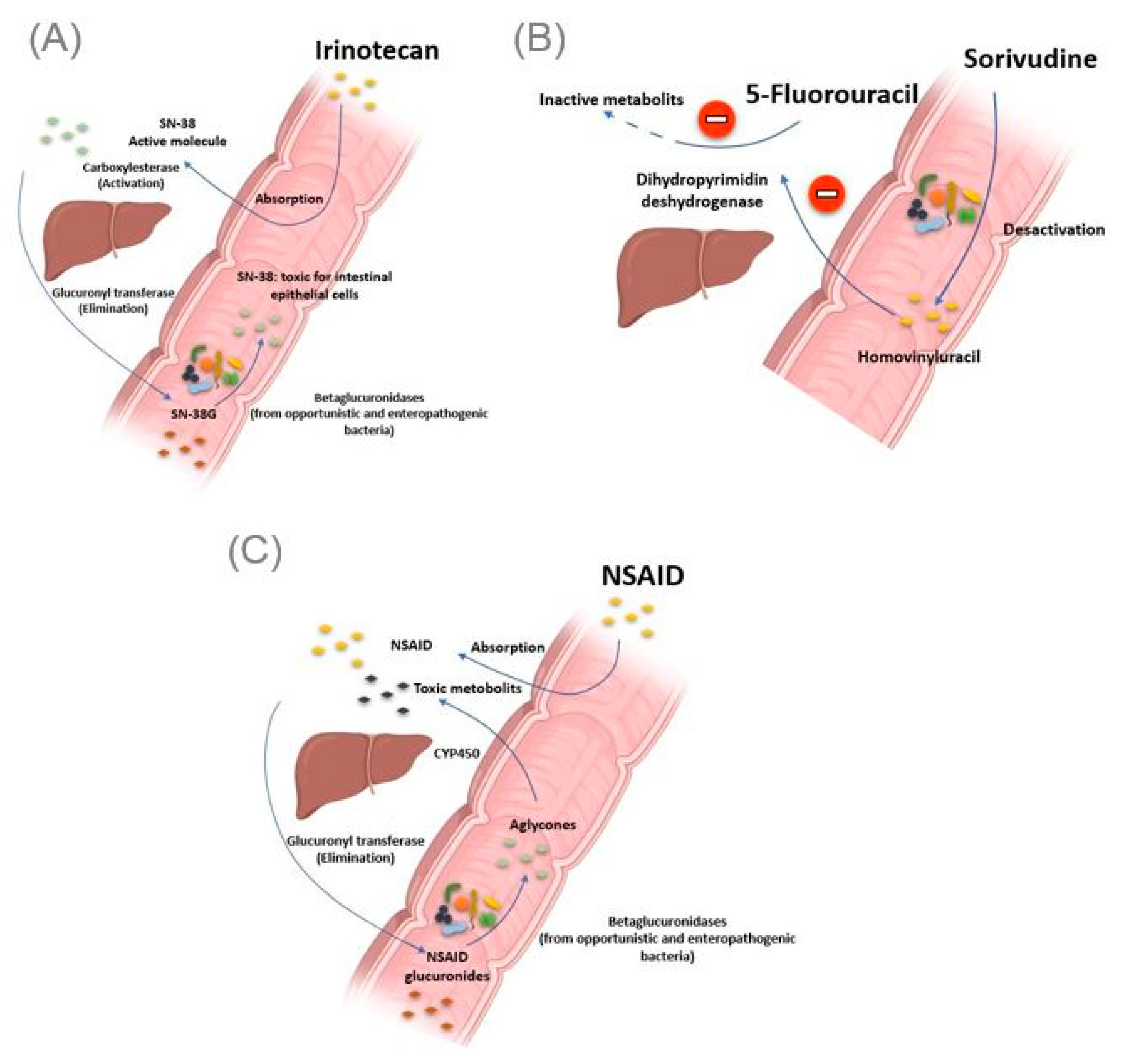
2.2.1. Irinotecan
2.2.2. Nonsteroidal Anti-Inflammatory Drugs
2.2.3. Impact of Non-Antibiotic Drugs on the Gut Microbiota
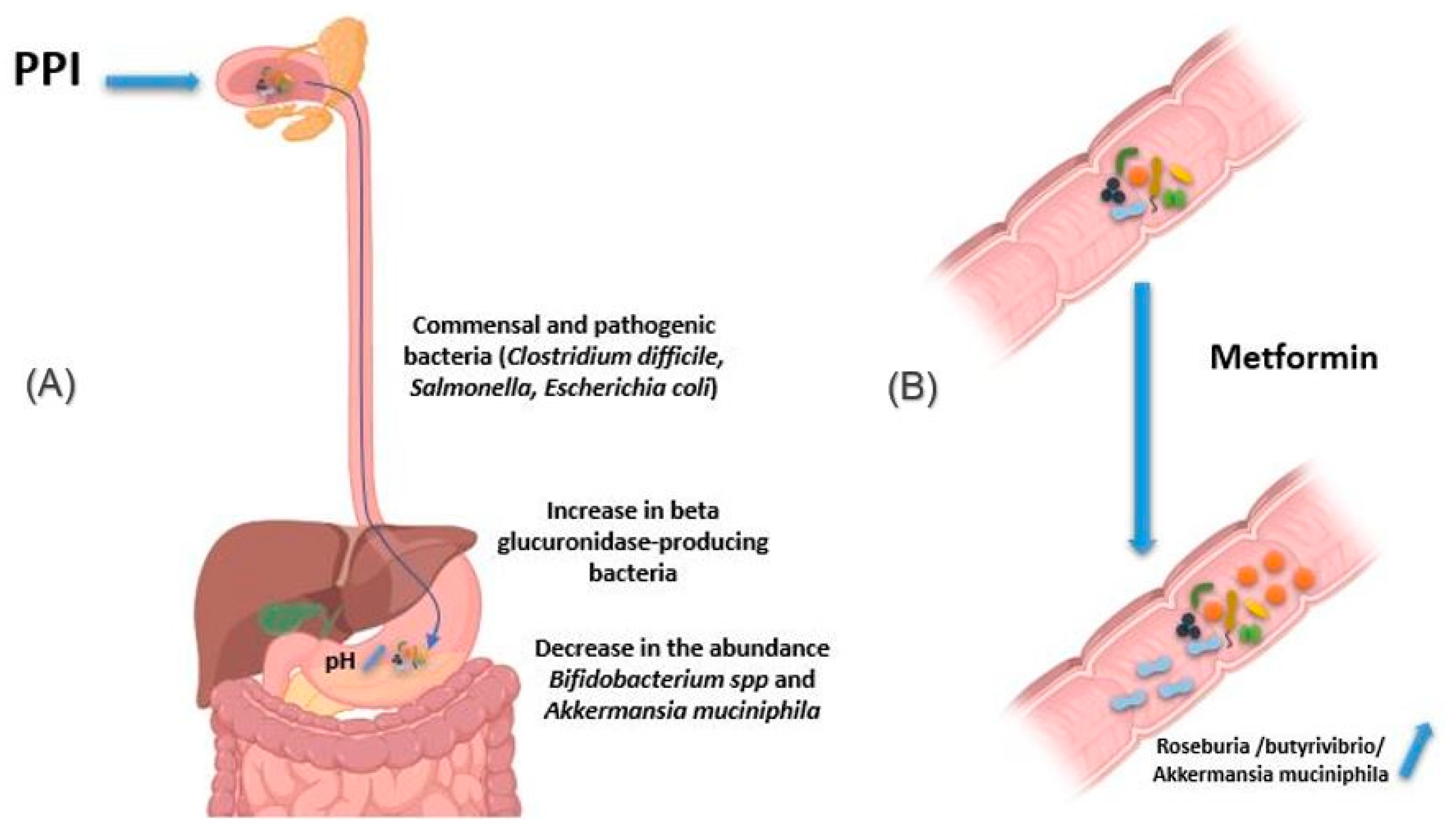
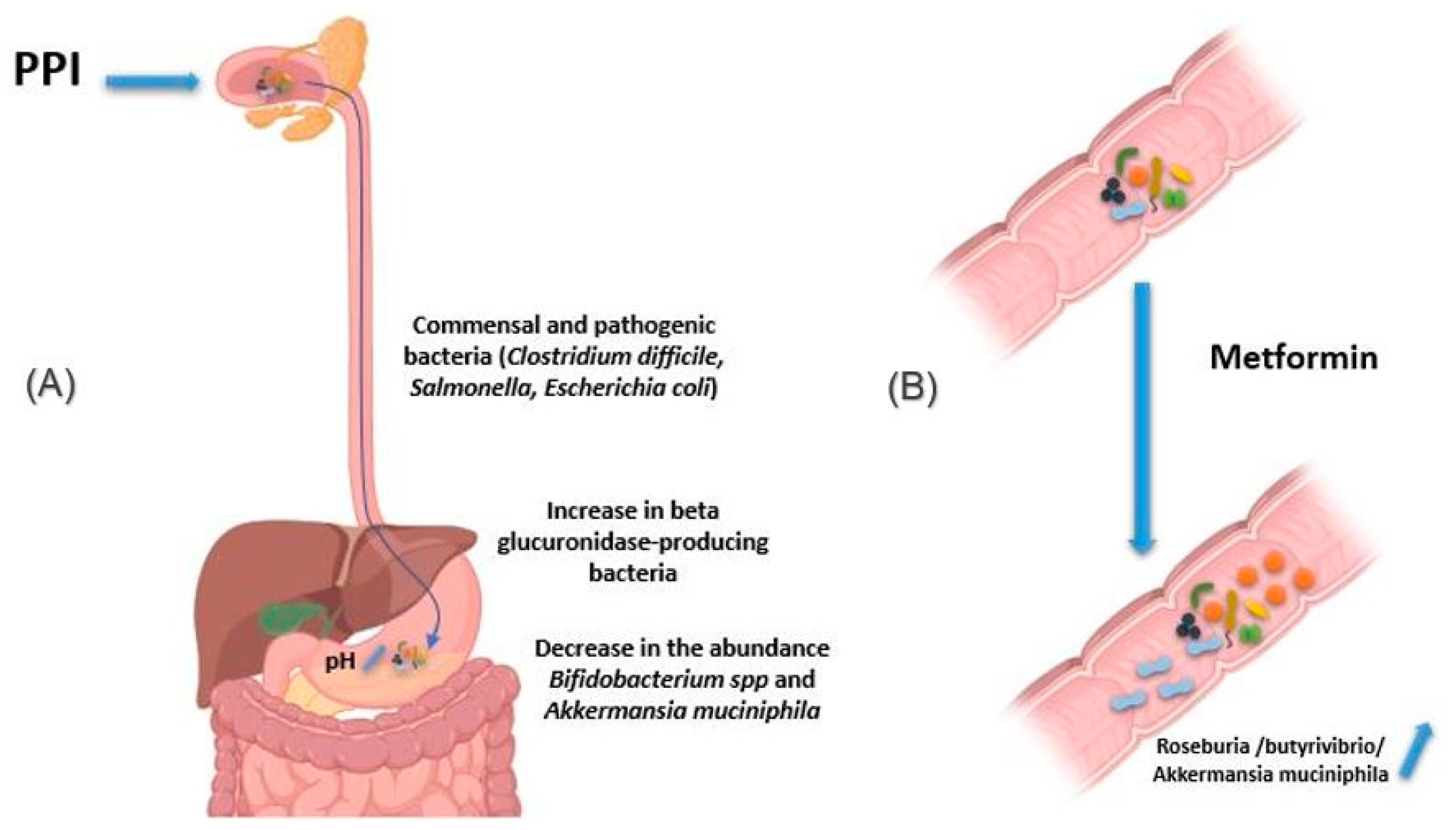
2.2.4. Proton Pomp Inhibitors
2.2.5. Metformin
3. Gut Microbiota—Drug Bidirectional Interaction
Methotrexate
4. Treatment Failure with TNF Alpha Inhibitors Due to Bacteria
5. Implementation of Pharmacomicrobiomics in Clinical Practice
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizkallah, M.R.; Saad, R.; Aziz, R.K. The Human Microbiome Project, personalized medicine and the birth of phar-macomicrobiomics. Curr. Pharm. Pers. Med. 2010, 8, 182–193. [Google Scholar]
- Birkett, D. Pharmacokinetics made easy: Therapeutic drug monitoring. Aust. Prescr. 1997, 20, 9–11. [Google Scholar] [CrossRef]
- Tange, S.M.; Grey, V.L.; Senécal, P.E. Therapeutic Drug Monitoring in Pediatrics: A Need for Improvement. J. Clin. Pharmacol. 1994, 34, 200–214. [Google Scholar] [CrossRef]
- Kalow, W. Pharmacogenomics: Historical Perspective and Current Status. Pharmacogenomics 2005, 311, 003–016. [Google Scholar] [CrossRef]
- Marshall, A. Getting the right drug into the right patient. Nat. Biotechnol. 1997, 15, 1249–1252. [Google Scholar] [CrossRef]
- Kalow, W.; Tang, B.-K.; Endrenyi, L. Hypothesis: Comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics 1998, 8, 283–289. [Google Scholar] [PubMed]
- Doestzada, M.; Vila, A.V.; Zhernakova, A.; Koonen, D.P.Y.; Weersma, R.K.; Touw, D.J.; Kuipers, F.; Wijmenga, C.; Fu, J. Pharmacomicrobiomics: A novel route towards personalized medicine? Protein Cell 2018, 9, 432–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Chen, L.; Shen, Z. Mechanisms of gastrointestinal microflora on drug metabolism in clinical practice. Saudi Pharm. J. 2019, 27, 1146–1156. [Google Scholar] [CrossRef]
- Feng, W.; Liu, J.; Ao, H.; Yue, S.; Peng, C. Targeting gut microbiota for precision medicine: Focusing on the efficacy and toxicity of drugs. Theranostics 2020, 10, 11278–11301. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Conlon, M.A.; Bird, A.R. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2015, 7, 17–44. [Google Scholar] [CrossRef]
- Kim, D.-H. Gut Microbiota-Mediated Drug-Antibiotic Interactions. Drug Metab. Dispos. 2015, 43, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Peppercorn, M.; Goldman, P. The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J. Pharmacol. Exp. Ther. 1972, 181, 555–562. [Google Scholar]
- Fuller, A.T. Is p-aminobenzenesulphonamide the active agent in protonsil therapy? Lancet 1937, 229, 194–198. [Google Scholar] [CrossRef]
- Pirmohamed, M. Warfarin: Almost 60 years old and still causing problems. Br. J. Clin. Pharmacol. 2006, 62, 509–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, E.S.; Gadomski, S.P.; McMaster, W.G.; Wilson, R.J.; Nelms, J.K.; Hocking, K.M.; Brophy, C.M. The influence of VKORC1 and CYP2C9 mutations on warfarin response after total hip and knee arthroplasty. J. Orthop. 2015, 12, S145–S151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Holford, N.; Ding, X.-L.; Shen, Z.-Y.; Huang, C.-R.; Zhang, H.; Zhang, J.-J.; Guo, Z.-N.; Xie, C.; Zhou, L.; et al. Theory-based pharmacokinetics and pharmacodynamics of S- and R-warfarin and effects on international normalized ratio: Influence of body size, composition and genotype in cardiac surgery patients. Br. J. Clin. Pharmacol. 2016, 83, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, L.; Liu, X.; Xiang, M.; Zhou, L.; Huang, C.; Shen, Z.; Miao, L. The gut microbes, Enterococcus and Escherichia-Shigella, affect the responses of heart valve replacement patients to the anticoagulant warfarin. Pharmacol. Res. 2020, 159, 104979. [Google Scholar] [CrossRef] [PubMed]
- MacLeod-Glover, N.; Mink, M.; Yarema, M.; Chuang, R. Digoxin toxicity Case for retiring its use in elderly patients? Can. Fam. Physician 2016, 62, 223–228. [Google Scholar]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and Manipulating Cardiac Drug Inactivation by the Human Gut Bacterium Eggerthella lenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Ugarte-Torres, A.; Gillrie, M.R.; Griener, T.P.; Church, D.L. Eggerthella lenta Bloodstream Infections Are Associated with Increased Mortality Following Empiric Piperacillin-Tazobactam (TZP) Monotherapy: A Population-based Cohort Study. Clin. Infect. Dis. 2018, 67, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiser, H.J.; Seim, K.L.; Balskus, E.P.; Turnbaugh, P.J. Mechanistic insight into digoxin inactivation byEggerthella lentaaugments our understanding of its pharmacokinetics. Gut Microbes 2014, 5, 233–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, N.; Bisanz, J.; Pandelia, M.-E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife 2018, 7, e33953. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.R.; Butler, V.P.; Neu, H.C.; Lindenbaum, J. Digoxin-inactivating bacteria: Identification in human gut flora. Science 1983, 220, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Jaiswal, S.K.; Dhoke, G.V.; Srivastava, G.N.; Sharma, A.K.; Sharma, V.K. Mechanistic and structural insight into promiscuity based metabolism of cardiac drug digoxin by gut microbial enzyme. J. Cell. Biochem. 2018, 119, 5287–5296. [Google Scholar] [CrossRef] [PubMed]
- Hashim, H.; Azmin, S.; Razlan, H.; Yahya, N.W.; Tan, H.J.; Manaf, M.R.; Ibrahim, N.M. Eradication of Helicobacter pylori infec-tion improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS ONE 2014, 9, e112330. [Google Scholar]
- Lahner, E.; Annibale, B.; Delle Fave, G. Systematic review: Helicobacter pylori infection and impaired drug absorp-tion. Aliment. Pharmacol. Ther. 2009, 29, 379–386. [Google Scholar] [CrossRef]
- Niehues, M.; Hensel, A. In-vitro interaction of L-dopa with bacterial adhesins of Helicobacter pylori: An explanation for clinicial differences in bioavailability? J. Pharm. Pharmacol. 2009, 61, 1303–1307. [Google Scholar] [CrossRef]
- Çamcı, G.; Oğuz, S. Association between Parkinson’s Disease and Helicobacter Pylori. J. Clin. Neurol. 2016, 12, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Pouncey, A.L.; Scott, A.J.; Alexander, J.L.; Marchesi, J.; Kinross, J. Gut microbiota, chemotherapy and the host: The influence of the gut microbiota on cancer treatment. Ecancermedicalscience 2018, 12, 868. [Google Scholar] [CrossRef] [Green Version]
- Erdman, S.E.; Poutahidis, T. Gut microbiota modulate host immune cells in cancer development and growth. Free. Radic. Biol. Med. 2017, 105, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal Bacteria Control Cancer Response to Therapy by Modulating the Tumor Microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Daillère, R.; Vétizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.R.; Nicholson, J.K.; Kinross, J.M. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356–365. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, A.; Voigt, W.; Jordan, K. Review: Chemotherapy-induced diarrhea: Pathophysiology, frequency and guideline-based management. Ther. Adv. Med. Oncol. 2010, 2, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodawara, T.; Higashi, T.; Negoro, Y.; Kamitani, Y.; Igarashi, T.; Watanabe, K.; Tsukamoto, H.; Yano, R.; Masada, M.; Iwasaki, H.; et al. The Inhibitory Effect of Ciprofloxacin on the β-Glucuronidase-mediated Deconjugation of the Irinotecan Metabolite SN-38-G. Basic Clin. Pharmacol. Toxicol. 2015, 118, 333–337. [Google Scholar] [CrossRef]
- Kong, R.; Liu, T.; Zhu, X.; Ahmad, S.; Williams, A.L.; Phan, A.T.; Zhao, H.; Scott, J.E.; Yeh, L.-A.; Wong, S.T. Old Drug New Use—Amoxapine and Its Metabolites as Potent Bacterial β-Glucuronidase Inhibitors for Alleviating Cancer Drug Toxicity. Clin. Cancer Res. 2014, 20, 3521–3530. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.W.; Tseng, C.H.; Tzeng, C.C.; Leu, Y.L.; Cheng, T.C.; Wang, J.Y.; Chang, J.M.; Lu, Y.C.; Cheng, C.M.; Chen, I.J.; et al. Pharma-cological inhibition of bacterial β-glucuronidase prevents irinotecan-induced diarrhea without impairing its antitumor effica-cy in vivo. Pharmacol. Res. 2019, 139, 41–49. [Google Scholar] [CrossRef]
- Nakamura, H.; Omori, S.; Kitada, K.; Mochida, A. Prevention of drug interactions and its countermeasure. J. Toxicol. Sci. 1994, 19, 89–93. [Google Scholar]
- Nakayama, H.; Kinouchi, T.; Kataoka, K.; Akimoto, S.; Matsuda, Y.; Ohnishi, Y. Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5-(E)-(2-bromovinyl)uracil that increases the level and toxicity of 5-fluorouracil. Pharmacogenetics 1997, 7, 35–43. [Google Scholar] [CrossRef]
- Wilkinson, E.M.; Ilhan, Z.E.; Herbst-Kralovetz, M.M. Microbiota–drug interactions: Impact on metabolism and efficacy of therapeutics. Maturitas 2018, 112, 53–63. [Google Scholar] [CrossRef]
- Diasio, R.B. Sorivudine and 5-fluorouracil; a clinically significant drug-drug interaction due to inhibition of dihydropyrimidine dehydrogenase. Br. J. Clin. Pharmacol. 1998, 46, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, K.; Umegaki, E.; Watanabe, T.; Yoda, Y.; Morita, E.; Murano, M.; Tokioka, S.; Arakawa, T. Present status and strategy of NSAIDs-induced small bowel injury. J. Gastroenterol. 2009, 44, 879–888. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Redinbo, M.R.; Saitta, K.S. Multiple NSAID-Induced Hits Injure the Small Intestine: Underlying Mechanisms and Novel Strategies. Toxicol. Sci. 2012, 131, 654–667. [Google Scholar] [CrossRef]
- Yauw, S.T.; Arron, M.; Lomme, R.M.; Broek, P.V.D.; Greupink, R.; Bhatt, A.P.; Redinbo, M.R.; Van Goor, H. Microbial Glucuronidase Inhibition Reduces Severity of Diclofenac-Induced Anastomotic Leak in Rats. Surg. Infect. 2018, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Dashnyam, P.; Mudududdla, R.; Hsieh, T.-J.; Lin, T.-C.; Lin, H.-Y.; Chen, P.-Y.; Hsu, C.-Y.; Lin, C.-H. β-Glucuronidases of opportunistic bacteria are the major contributors to xenobiotic-induced toxicity in the gut. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ervin, S.M.; Hanley, R.P.; Lim, L.; Walton, W.G.; Pearce, K.H.; Bhatt, A.P.; James, L.I.; Redinbo, M.R. Targeting Regorafenib-Induced Toxicity through Inhibition of Gut Microbial β-Glucuronidases. ACS Chem. Biol. 2019, 14, 2737–2744. [Google Scholar] [CrossRef]
- Yang, B.; Liu, H.; Yang, J.; Gupta, V.K.; Jiang, Y. New insights on bioactivities and biosynthesis of flavonoid glycosides. Trends Food Sci. Technol. 2018, 79, 116–124. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Wright, B.; Spencer, J.P.; Lovegrove, J.A.; Gibbins, J.M. Insights into dietary flavonoids as molecular templates for the design of anti-platelet drugs. Cardiovasc. Res. 2012, 97, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedberg, D.E.; Lebwohl, B.; Abrams, J.A. The Impact of Proton Pump Inhibitors on the Human Gastrointestinal Microbiome. Clin. Lab. Med. 2014, 34, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Imhann, F.; Bonder, M.J.; Vila, A.V.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.; Cenit, M.C.; Harmsen, H.J.M.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dial, S.; Alrasadi, K.; Manoukian, C.; Huang, A.; Menzies, D. Risk of Clostridium difficile diarrhea among hospital inpatients prescribed proton pump inhibitors: Cohort and case-control studies. Can. Med. Assoc. J. 2004, 171, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, G.; Zaccari, P.; Rocco, G.; Scalese, G.; Panetta, C.; Porowska, B.; Pontone, S.; Severi, C. Proton pump inhibitors and dysbiosis: Current knowledge and aspects to be clarified. World J. Gastroenterol. 2019, 25, 2706–2719. [Google Scholar] [CrossRef] [PubMed]
- Blackler, R.W.; Motta, J.-P.; Manko, A.; Workentine, M.; Bercik, P.; Surette, M.G.; Wallace, J.L. Hydrogen sulphide protects against NSAID-enteropathy through modulation of bile and the microbiota. Br. J. Pharmacol. 2014, 172, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.A.; Collier, F.; Mohebbi, M.; Stuart, A.L.; Loughman, A.; Pasco, J.A.; Jacka, F.N. Obesity, Akkermansia muciniphila, and Proton Pump Inhibitors: Is there a Link? Obes. Res. Clin. Pr. 2020, 14, 524–530. [Google Scholar] [CrossRef]
- Wallace, J.L.; Syer, S.; Denou, E.; de Palma, G.; Vong, L.; McKnight, W.; Jury, J.; Bolla, M.; Bercik, P.; Collins, S.M.; et al. Proton Pump Inhibitors Exacerbate NSAID-Induced Small Intestinal Injury by Inducing Dysbiosis. Gastroenterology 2011, 141, 1314–1322.e5. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Lutz, O.M. Pharmacology of metformin—An update. Eur. J. Pharmacol. 2019, 865, 172782. [Google Scholar] [CrossRef]
- Kim, C.H. Microbiota or short-chain fatty acids: Which regulates diabetes? Cell. Mol. Immunol. 2018, 15, 88–91. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.R.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population in-duced by metformin treatment improves glucose homeostasis in dietinduced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatmentnaive type 2 diabetes, contributing to the thera-peutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- De La Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated with Higher Relative Abundance of Mucin-DegradingAkkermansia muciniphilaand Several Short-Chain Fatty Acid–Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 63, 1513–1521. [Google Scholar] [CrossRef]
- Harsch, I.A.; Konturek, P.C. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med. Sci. 2018, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Romero, S.; Hereu, M.; Atienza, L.; Casas, J.; Jáuregui, O.; Amézqueta, S.; DaSilva, G.; Medina, I.; Nogués, M.R.; Romeu, M.; et al. Mechanistically different effects of fat and sugar on insulin resistance, hypertension, and gut microbiota in rats. Am. J. Physiol. Metab. 2018, 314, E552–E563. [Google Scholar] [CrossRef]
- Dunn, C.J.; Peters, D.H. Metformin. Drugs 1995, 49, 721–749. [Google Scholar] [CrossRef]
- Zhou, B.; Xia, X.; Wang, P.; Chen, S.; Yu, C.; Huang, R.; Zhang, R.; Wang, Y.; Lu, L.; Yuan, F.; et al. Induction and Amelioration of Methotrexate-Induced Gastrointestinal Toxicity are Related to Immune Response and Gut Microbiota. EBioMedicine 2018, 33, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.; MacGregor, A.; Carding, S.R. Drug-microbiota interactions and treatment response: Relevance to rheumatoid arthritis. AIMS Microbiol. 2018, 4, 642–654. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Goodman, S.M.; Cronstein, B.N.; Bykerk, V.P. Outcomes related to methotrexate dose and route of administration in patients with rheumatoid arthritis: A systematic literature review. Clin. Exp. Rheumatol. 2014, 33, 272–278. [Google Scholar]
- Scher, J.U.; Nayak, R.R.; Ubeda, C.; Turnbaugh, P.J.; Abramson, S.B. Pharmacomicrobiomics in inflammatory arthritis: Gut microbiome as modulator of therapeutic response. Nat. Rev. Rheumatol. 2020, 16, 282–292. [Google Scholar] [CrossRef]
- Halilova, K.I.; Brown, E.E.; Morgan, S.L.; Bridges, S.L.; Hwang, M.-H.; Arnett, N.K.; Danila, M.I. Markers of Treatment Response to Methotrexate in Rheumatoid Arthritis: Where Do We Stand? Int. J. Rheumatol. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Angelis-Stoforidis, P.; Vajda, F.J.; Christophidis, N. Methotrexate polyglutamate levels in circulating erythrocytes and pol-ymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin. Exp. Rheumatol. 1999, 17, 313–320. [Google Scholar]
- Dervieux, T.; Furst, D.; Lein, D.O.; Capps, R.; Smith, K.; Walsh, M.; Kremer, J. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004, 50, 2766–2774. [Google Scholar] [CrossRef]
- Glaser, C.; Rieg, S.; Wiech, T.; Scholz, C.; Endres, D.; Stich, O.; Hasselblatt, P.; Geißdörfer, W.; Bogdan, C.; Serr, A.; et al. Whipple’s disease mimicking rheumatoid arthritis can cause misdiagnosis and treatment failure. Orphanet J. Rare Dis. 2017, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneitz, C.; Suerbaum, S.; Beer, M.; Müller, J.; Jahns, R.; Tony, H. Exacerbation of Whipple’s disease associated with infliximab treatment. Scand. J. Rheumatol. 2005, 34, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.M.; Pasquau, F.; Galipienso, N.; Valero, B.; Navarro, A.; Martinez, A.; Rosas, J.; Gutierrez, A.; Sánchez-Martínez, R. Whipple’s disease diagnosed during anti-tumor necrosis factor alpha treatment: Two case reports and review of the literature. J. Med. Case Rep. 2015, 9, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bureš, J.; Kopáčová, M.; Douda, T.; Bártová, J.; Tomš, J.; Rejchrt, S.; Tachecí, I. Whipple’s Disease: Our Own Experience and Review of the Literature. Gastroenterol. Res. Pract. 2013, 2013, 478349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenollar, F.; Lagier, J.-C.; Raoult, D. Tropheryma whipplei and Whipple’s disease. J. Infect. 2014, 69, 103–112. [Google Scholar] [CrossRef]
- Lepidi, H.; Fenollar, F.; Dumler, J.S.; Gauduchon, V.; Chalabreysse, L.; Bammert, A.; Bonzi, M.F.; Thivolet-Bejui Vandenesch Raoult, D. Cardiac valves in patients with Whipple endocarditis: Microbiological, molecular, quantitative histologic and im-munohistochemical studies of 5 patients. J. Infect. Dis. 2004, 190, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marth, T. Complicated Whipple’s disease and endocarditis following tumor necrosis factor inhibitors. World J. Cardiol. 2014, 6, 1278–1284. [Google Scholar] [CrossRef]
- Marth, T. Systematic review: Whipple’s disease (Tropheryma whippleiinfection) and its unmasking by tumour necrosis factor inhibitors. Aliment. Pharmacol. Ther. 2015, 41, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.; Kaakoush, N.; Walsh, A.J.; Bogaerde, J.V.D.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Juul, F.E.; Garborg, K.; Bretthauer, M.; Skudal, H.; Øines, M.N.; Wiig, H.; Rose, Ø.; Seip, B.; Lamont, J.T.; Midtvedt, T.; et al. Fecal Microbiota Transplantation for Primary Clostridium difficile Infection. N. Engl. J. Med. 2018, 378, 2535–2536. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wiesnoski, D.H.; Helmink, B.A.; Gopalakrishnan, V.; Choi, K.; Dupont, H.L.; Jiang, Z.-D.; Abu-Sbeih, H.; Sanchez, C.A.; Chang, C.-C.; et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 2018, 24, 1804–1808. [Google Scholar] [CrossRef]
- Chang, C.W.; Lee, H.C.; Li, L.H.; Chiang Chiau, J.S.; Wang, T.E.; Chuang, W.H.; Chen, M.J.; Wang, H.Y.; Shih, S.C.; Liu, C.Y.; et al. Fecal microbiota transplantation prevents intestinal injury, upregulation of toll-like receptors, and 5 fluoroura-cil/oxaliplatin-induced toxicity in colorectal cancer. Int. J. Mol. Sci. 2020, 21, 386. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.T.; Cai, H.F.; Wang, Z.H.; Xu, J.; Fang, J.Y. Systematic review with meta-analysis: Long-term outcomes of faecal microbiota transplantation for Clostridium difficile infection. Aliment. Pharmacol. Ther. 2016, 43, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic review with meta-analysis: Faecal micro-biota transplantation for the induction of remission for active ulcerative colitis. Aliment. Pharmacol. Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Microbs | Body Site | Effects | References |
|---|---|---|---|---|
| Drug Effect Influenced by Bacteria | ||||
| Sulfasalazine | Bacteria possessing azoreductase enzymes | Gut | Cleavage into its two active metabolites, Salazopyrin and 5-amine salicylic acid | Peppercorn MA and Goldman P, 1972 |
| Warfarin | Bacteroides, Escherichia–Shigella and Klebsiella | Gut | weak response to the drug | Wang L et al., 2020 |
| Enterococcus | Gut | High response to the drug | ||
| Digoxin | Eggerthella lenta | Gut | reduction of digoxin to its inactive metabolite, dihydro-digoxin | Haiser HJ et al., 2014; Koppel N et al., 2018 |
| Levodopa | Helicobacter pylori | Stomach | decreased drug absorption | Hashim H et al., 2014 |
| Cyclophosphamide (CTX) | Enterococcus hirae, Lactobacillus johnsonii, Lactobacillus murinus | Mesenteric lymph nodes and the spleen | Enhancement of the antitumor adaptive immunological response to CTX | Viaud S et al., 2013; Daillère et al., 2016 |
| CTLA-4 checkpoint inhibitors | Bacteroides fragilis | Gut | Restore the response to the treatment | Vétizou M et al., 2015 |
| Anti PD-1 | Akkermentia muciniphila, Collinsella aerofaciens, Enterococcus faecium, Ruminococcaceae family, Bifidobacterium spp. | Gut | Enhanced response to treatment | Gopalakrishnan V et al., 2018; Matson V et al., 2018; Routy B et al., 2017 |
| Gemcitabine | Mycoplasma hyorhinis, bacteria belonging to the Gammaproteobacteria, Escherichia coli | Tumor tissue | Gemcitabine resistance | Galler et al., 2017; Lehouritis P et al., 2015 |
| Irinotecan | Opportunistic or enterohepatic bacteria possessing β-glucuronidases enzymes | Gut | Production of toxic metabolites responsible for diarrhea | Stein A et al., 2010 |
| NSAIDs | Gut | Production of toxic metabolites responsible for mucosal damage in the small intestine | Higuchi et al., 2009; Boelsterli UA et al., 2013 | |
| Bacteria abundance influenced by drugs | ||||
| Proton pump inhibitors | Clostridium difficile, Salmonella, diarrheagenic Escherichia coli and beta glucuronidase-producing bacteria | Gut | Increased bacteria | Dial et al., 2004; Bruno G et al., 2019; Blackler RW et al., 2015; Davis JA et al., 2020; Wallace JL et al., 2011 |
| Bifidobacterium spp. and Akkermentia muciniphila | Gut | Decreased bacteria | ||
| Metformin | Roseburia, butyrivibrio genera and Akkermentia muciniphila | Increased bacteria, responsible for better epithelial permeability and improvement in glucose and lipid metabolism | Forslund K et al., 2015; Shin NR et al., 2014; Wu H et al., 2017 | |
| Bidirectional effect | ||||
| Methotrexate (MTX) | Enterobacterial group, Ruminococcaceae, Bacteroidetes phyla and Bacteroides fragilis | Gut | Decreased bacteria | Ramos-Romero S et al., 2018; Zhou B et al., 2018 |
| Lachnospiraceae family | Gut | Increased bacteria | ||
| Prevotella maculosa | Gut | Enhancement of the response to the treatment | Zhang et al., 2015 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannachi, N.; Camoin-Jau, L. Drug Response Diversity: A Hidden Bacterium? J. Pers. Med. 2021, 11, 345. https://doi.org/10.3390/jpm11050345
Hannachi N, Camoin-Jau L. Drug Response Diversity: A Hidden Bacterium? Journal of Personalized Medicine. 2021; 11(5):345. https://doi.org/10.3390/jpm11050345
Chicago/Turabian StyleHannachi, Nadji, and Laurence Camoin-Jau. 2021. "Drug Response Diversity: A Hidden Bacterium?" Journal of Personalized Medicine 11, no. 5: 345. https://doi.org/10.3390/jpm11050345
APA StyleHannachi, N., & Camoin-Jau, L. (2021). Drug Response Diversity: A Hidden Bacterium? Journal of Personalized Medicine, 11(5), 345. https://doi.org/10.3390/jpm11050345