
Transmembrane Chloride Intracellular Channel 1 (tmCLIC1) as a Potential Biomarker for Personalized Medicine

{kind=link}
Abstract
:1. CLIC Proteins, a Focus on CLIC1
2. CLIC1 in Non-Pathological States
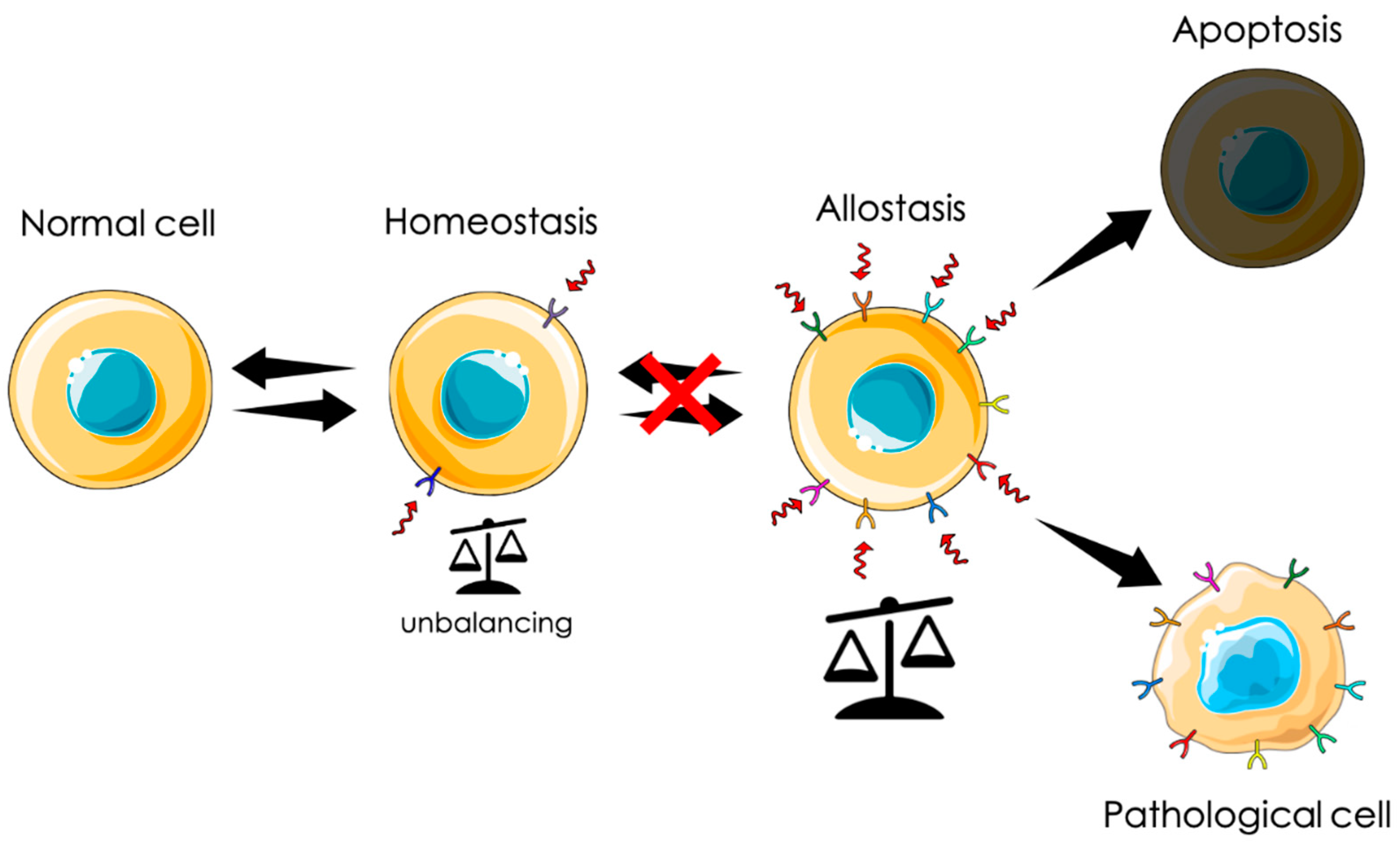
3. Activation of Transitory Allostasis through CLIC1 Function
4. CLIC1 during Chronic Allostasis
4.1. CLIC1 in Solid Tumors
4.2. CLIC1 in Neurodegenerative Processes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Littler, D.R.; Harrop, S.J.; Goodchild, S.; Phang, J.M.; Mynott, A.V.; Jiang, L.; Valenzuela, S.; Mazzanti, M.; Brown, L.; Breit, S.N.; et al. The enigma of the CLIC proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett. 2010, 584, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Harrop, S.J.; DeMaere, M.; Fairlie, W.; Reztsova, T.; Valenzuela, S.; Mazzanti, M.; Tonini, R.; Qiu, M.R.; Jankova, L.; Warton, K.; et al. Crystal Structure of a Soluble Form of the Intracellular Chloride Ion Channel CLIC1 (NCC27) at 1.4-Å Resolution. J. Biol. Chem. 2001, 276, 44993–45000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littler, D.R.; Harrop, S.J.; Fairlie, W.; Brown, L.; Pankhurst, G.J.; Pankhurst, S.; DeMaere, M.; Campbell, T.; Bauskin, A.R.; Tonini, R.; et al. The Intracellular Chloride Ion Channel Protein CLIC1 Undergoes a Redox-controlled Structural Transition. J. Biol. Chem. 2004, 279, 9298–9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulk, B.M.; Kapadia, S.; Edwards, J.C. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am. J. Physiol. Cell Physiol. 2002, 282, C1103–C1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warton, K.; Tonini, R.; Fairlie, W.D.; Matthews, J.M.; Valenzuela, S.M.; Qiu, M.R.; Wu, W.M.; Pankhurst, S.; Bauskin, A.R.; Harrop, S.J.; et al. Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J. Biol. Chem. 2002, 277, 26003–26011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J.; Stein, V.; Weinreich, F.; Zdebik, A.A. Molecular Structure and Physiological Function of Chloride Channels. Physiol. Rev. 2002, 82, 503–568. [Google Scholar] [CrossRef]
- Ueno, Y.; Ozaki, S.; Umakoshi, A.; Yano, H.; Choudhury, M.E.; Abe, N.; Sumida, Y.; Kuwabara, J.; Uchida, R.; Islam, A.; et al. Chloride intracellular channel protein 2 in cancer and non-cancer human tissues: Relationship with tight junctions. Tissue Barriers 2019, 7, 1593775. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wang, Z.; Dong, M.; Wu, D.; Liao, S.; Li, X. Chloride intracellular channel protein 2: Prognostic marker and correlation with PD-1/PD-L1 in breast cancer. Aging 2020, 12, 17305–17327. [Google Scholar] [CrossRef]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins Struct. Funct. Bioinform. 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhang, S.; Wen, X.; Cao, H.; Gao, Y. Prognostic value of CLIC3 mRNA overexpression in bladder cancer. PeerJ 2020, 8, e8348. [Google Scholar] [CrossRef]
- Patel, S.H.; Edwards, M.J.; Ahmad, S.A. Intracellular Ion Channels in Pancreas Cancer. Cell. Physiol. Biochem. 2019, 53, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, I.R.; Rainero, E.; Mitchell, L.E.; Berghe, P.V.V.D.; Speirs, C.; Dozynkiewicz, M.A.; Chaudhary, S.; Kalna, G.; Edwards, J.; Timpson, P.; et al. CLIC3 controls recycling of late endosomal MT1-MMP and dictates invasion and metastasis in breast cancer. J. Cell Sci. 2014, 127, 3893–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Fernaud, J.R.; Ruengeler, E.; Casazza, A.; Neilson, L.J.; Pulleine, E.; Santi, A.; Ismail, S.; Lilla, S.; Dhayade, S.; MacPherson, I.R.; et al. Secreted CLIC3 drives cancer progression through its glutathione-dependent oxidoreductase activity. Nat. Commun. 2017, 8, 14206. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Fernández, R.; Coll, R.C.; Kearney, J.; Breit, S.; O’Neill, L.A. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017, 292, 12077–12087. [Google Scholar] [CrossRef] [Green Version]
- Kagiali ZC, U.; Saner, N.; Akdag, M.; Sanal, E.; Degirmenci, B.S.; Mollaoglu, G.; Ozlu, N. CLIC4 and CLIC1 bridge plasma membrane and cortical actin network for a successful cytokinesis. Life Sci. Alliance 2019, 3, e201900558. [Google Scholar] [CrossRef] [PubMed]
- Tavasoli, M.; Al-Momany, A.; Wang, X.; Li, L.; Edwards, J.C.; Ballermann, B.J. Both CLIC4 and CLIC5A activate ERM proteins in glomerular endothelium. Am. J. Physiol. Renal Physiol. 2016, 311, F945–F957. [Google Scholar] [CrossRef]
- Valenzuela, S.; Martin, D.K.; Por, S.B.; Robbins, J.M.; Warton, K.; Bootcov, M.; Schofield, P.; Campbell, T.J.; Breit, S.N. Molecular Cloning and Expression of a Chloride Ion Channel of Cell Nuclei. J. Biol. Chem. 1997, 272, 12575–12582. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Ashley, R.H. Redox Regulation of CLIC1 by Cysteine Residues Associated with the Putative Channel Pore. Biophys. J. 2006, 90, 1628–1638. [Google Scholar] [CrossRef] [Green Version]
- Tonini, R.; Ferroni, A.; Valenzuela, S.; Warton, K.; Campbell, T.; Breit, S.N.; Mazzanti, M. Functional characterization of the NCC27 nuclear protein in stable transfected CHO-K1 cells. FASEB J. 2000, 14, 1171–1178. [Google Scholar] [CrossRef]
- Al Khamici, H.; Hossain, K.R.; Cornell, B.A.; Valenzuela, S.M. Investigating Sterol and Redox Regulation of the Ion Channel Activity of CLIC1 Using Tethered Bilayer Membranes. Membranes 2016, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.R.; Jiang, L.; Matthaei, K.I.; Schoenwaelder, S.M.; Kuffner, T.; Mangin, P.; Joseph, J.E.; Low, J.; Connor, D.; Valenzuela, S.M.; et al. Generation and characterization of mice with null mutation of the chloride intracellular channel 1 gene. Genesis 2010, 48, 127–136. [Google Scholar] [CrossRef]
- Liu, B.; Billington, C.K.; Henry, A.P.; Bhaker, S.K.; Kheirallah, A.K.; Swan, C.; Hall, I. Chloride intracellular channel 1 (CLIC1) contributes to modulation of cyclic AMP-activated whole-cell chloride currents in human bronchial epithelial cells. Physiol. Rep. 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Khamici, H.; Brown, L.J.; Hossain, K.R.; Hudson, A.L.; Sinclair-Burton, A.A.; Ng, J.P.M.; Daniel, E.L.; Hare, J.E.; Cornell, B.A.; Curmi, P.M.G.; et al. Members of the Chloride Intracellular Ion Channel Protein Family Demonstrate Glutaredoxin-Like Enzymatic Activity. PLoS ONE 2015, 10, e115699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, J.C.; Joughin, B.A.; Van De Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Hu, P.; Li, Y.; Win-Shwe, T.-T.; Li, C. Ion Imbalance Is Involved in the Mechanisms of Liver Oxidative Damage in Rats Exposed to Glyphosate. Front. Physiol. 2017, 8, 1083. [Google Scholar] [CrossRef] [PubMed]
- Peretti, M.; Raciti, F.M.; Carlini, V.; Verduci, I.; Sertic, S.; Barozzi, S.; Garre’, M.; Pattarozzi, A.; Daga, A.; Barbieri, F.; et al. Mutual Influence of ROS, pH, and CLIC1 Membrane Protein in the Regulation of G1–S Phase Progression in Human Glioblastoma Stem Cells. Mol. Cancer Ther. 2018, 17, 2451–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Grueso, M.J.; González-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.; Muntané, J.; Bárcena, J.; Padilla, C. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 2019, 21, 101049. [Google Scholar] [CrossRef]
- Jiang, L.; Salao, K.; Li, H.; Rybicka, J.M.; Yates, R.M.; Luo, X.W.; Shi, X.X.; Kuffner, T.; Tsai, V.W.-W.; Husaini, Y.; et al. Intracellular chloride channel protein CLIC1 regulates macrophage functions via modulation of phagosomal acidification. J. Cell Sci. 2012, 125, 5479–5488. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Łuczak, K.; Balcerczyk, A.; Soszynski, M.; Bartosz, G.; Uczak, K. Low concentration of oxidant and nitric oxide donors stimulate proliferation of human endothelial cells in vitro. Cell Biol. Int. 2004, 28, 483–486. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling. Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Tung, J.J.; Kitajewski, J. Chloride intracellular channel 1 functions in endothelial cell growth and migration. J. Angiogenes Res. 2010, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhu, J.; Hu, X.; Wang, C.; Lu, D.; Gong, C.; Yang, J.; Zong, L. CLIC1 Inhibition Attenuates Vascular Inflammation, Oxidative Stress, and Endothelial Injury. PLoS ONE 2016, 11, e0166790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milton, R.H.; Abeti, R.; Averaimo, S.; DeBiasi, S.; Vitellaro, L.; Jiang, L.; Curmi, P.M.G.; Breit, S.N.; Duchen, M.R.; Mazzanti, M. CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J. Neurosci. 2008, 28, 11488–11499. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Dong, Q.; Zhang, B.; Wang, X.; Ye, B.; Zhang, F.; Song, X.; Gao, G.; Mu, J.; Wang, Z.; et al. Chloride intracellular channel 1 (CLIC1) is activated and functions as an oncogene in pancreatic cancer. Med. Oncol. 2015, 32, 171. [Google Scholar] [CrossRef]
- Lee, J.R.; Lee, J.Y.; Kim, H.J.; Hahn, M.J.; Kang, J.S.; Cho, H. The inhibition of chloride intracellular channel 1 enhances Ca(2+) and reactive oxygen species signaling in A549 human lung cancer cells. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar]
- Yu, W.; Cui, R.; Qu, H.; Liu, C.; Deng, H.; Zhang, Z. Expression and prognostic value of CLIC1 in epithelial ovarian cancer. Exp. Ther. Med. 2018, 15, 4943–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Li, B.-P.; Mao, Y.-T.; Wang, Z.; Chen, Y.-Y.; Wang, Y.; Zhai, C.-Y.; Shi, B.; Liu, S.-Y.; Liu, J.-L.; Chen, J.-Q. CLIC1 Promotes the Progression of Gastric Cancer by Regulating the MAPK/AKT Pathways. Cell. Physiol. Biochem. 2018, 46, 907–924. [Google Scholar] [CrossRef]
- Meng, M.; Wang, H.B.; Song, S.Z.; Sun, R.; Lin, Y.; Lin, S. CLIC1 facilitates cancer associated fibroblast activation and gastric cancer progression via integrins/NF-kappaB pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 320, G836. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, C.; Yu, P.; Tang, B.; Liu, T.; Cui, H.; Xu, J. Regulation of colon cancer cell migration and invasion by CLIC1-mediated RVD. Mol. Cell. Biochem. 2012, 365, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Brescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional Role of CLIC1 Ion Channel in Glioblastoma-Derived Stem/Progenitor Cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Li, M.; Wu, X.; Zhang, L.; Wu, W.; Ding, Q.; Weng, H.; Wang, X.; Liu, Y. CLIC1 overexpression is associated with poor prognosis in gallbladder cancer. Tumour Biol. 2015, 36, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef] [Green Version]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Novarino, G.; Fabrizi, C.; Tonini, R.; Denti, M.A.; Malchiodi-Albedi, F.; Lauro, G.; Sacchetti, B.; Paradisi, S.; Ferroni, A.; Curmi, P.M.; et al. Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J. Neurosci. 2004, 24, 5322–5330. [Google Scholar] [CrossRef] [Green Version]
- Paradisi, S.; Matteucci, A.; Fabrizi, C.; Denti, M.A.; Abeti, R.; Breit, S.N.; Malchiodi-Albedi, F.; Mazzanti, M. Blockade of chloride intracellular ion channel 1 stimulates Aβ phagocytosis. J. Neurosci. Res. 2008, 86, 2488–2498. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Intracellular ion channel CLIC1: Involvement in microglia-mediated beta-amyloid peptide(1-42) neurotoxicity. Neurochem. Res. 2013, 38, 1801–1808. [Google Scholar] [CrossRef]
- Carlini, V.; Verduci, I.; Cianci, F.; Cannavale, G.; Fenoglio, C.; Galimberti, D.; Mazzanti, M. CLIC1 Protein Accumulates in Circulating Monocyte Membrane during Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 1484. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.B.; Kauwe, J.S.K. Predicting Clinical Dementia Rating Using Blood RNA Levels. Genes 2020, 11, 706. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianci, F.; Verduci, I. Transmembrane Chloride Intracellular Channel 1 (tmCLIC1) as a Potential Biomarker for Personalized Medicine. J. Pers. Med. 2021, 11, 635. https://doi.org/10.3390/jpm11070635
Cianci F, Verduci I. Transmembrane Chloride Intracellular Channel 1 (tmCLIC1) as a Potential Biomarker for Personalized Medicine. Journal of Personalized Medicine. 2021; 11(7):635. https://doi.org/10.3390/jpm11070635
Chicago/Turabian StyleCianci, Francesca, and Ivan Verduci. 2021. "Transmembrane Chloride Intracellular Channel 1 (tmCLIC1) as a Potential Biomarker for Personalized Medicine" Journal of Personalized Medicine 11, no. 7: 635. https://doi.org/10.3390/jpm11070635
APA StyleCianci, F., & Verduci, I. (2021). Transmembrane Chloride Intracellular Channel 1 (tmCLIC1) as a Potential Biomarker for Personalized Medicine. Journal of Personalized Medicine, 11(7), 635. https://doi.org/10.3390/jpm11070635