
JDSNMF: Joint Deep Semi-Non-Negative Matrix Factorization for Learning Integrative Representation of Molecular Signals in Alzheimer’s Disease


Abstract
:1. Introduction
- 1.
- It introduces a hierarchical non-linear unsupervised learning model for capturing complex information from multi-omics data.
- 2.
- It introduces a flexible integration model for multiple data which does not require matching samples.
- 3.
- We compared the feature extraction performance of JDSNMF using sample-matched simulated data and feature-matched AD data.
- 4.
2. Related Work
2.1. Non-Negative Matrix Factorization
2.2. Joint NMF
2.3. Semi-NMF
2.4. Deep Semi-NMF
3. Proposed Method
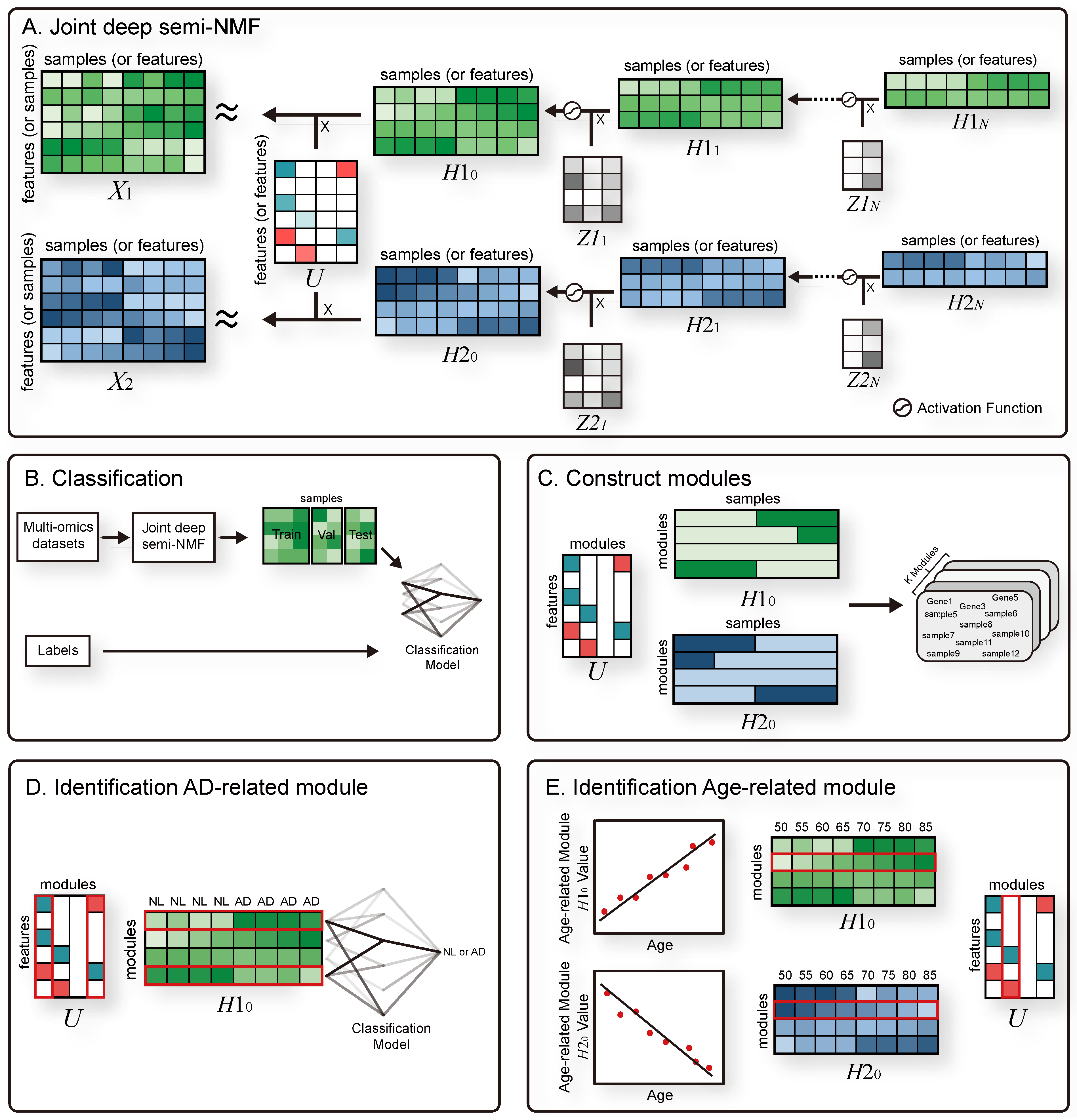
3.1. Joint Deep Semi-NMF
3.2. Constructing Modules
4. Results
4.1. Dataset
4.2. Hyperparameters and Settings for Experiments
4.3. Sample-Matched Simulated Dataset Experiments
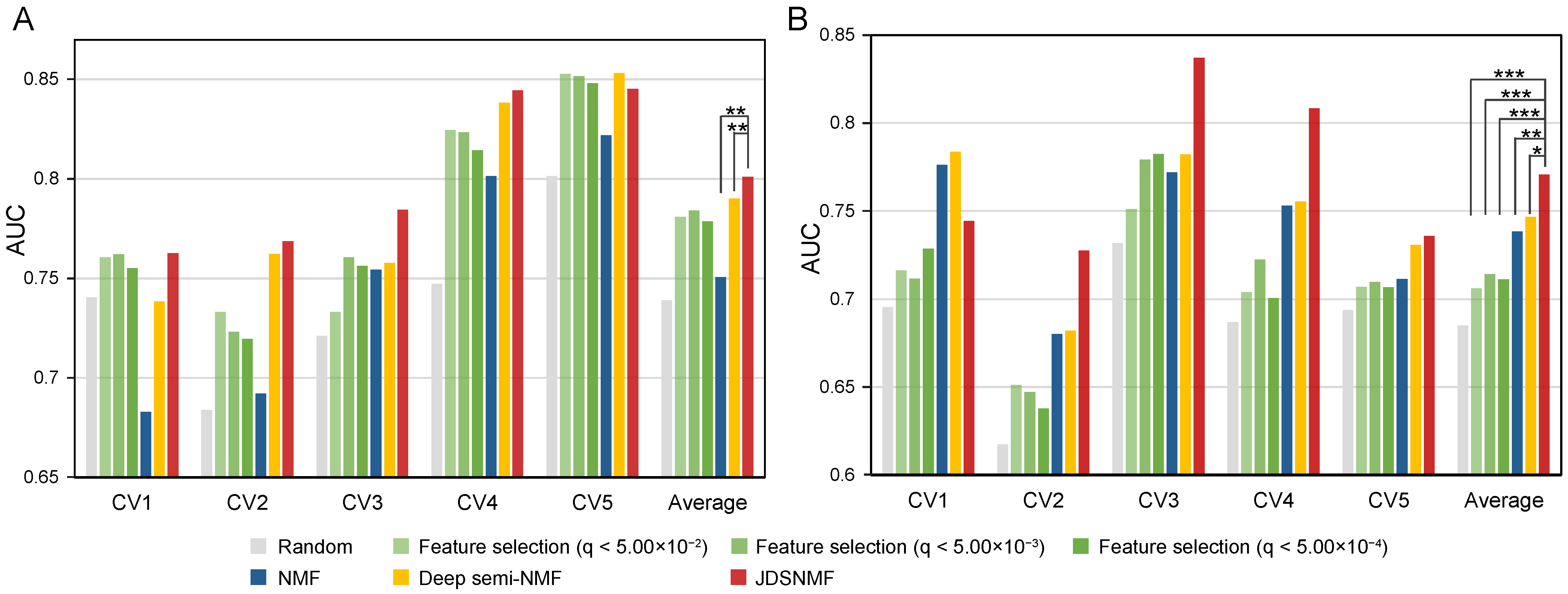
4.4. Feature-Matched Real Dataset Experiments
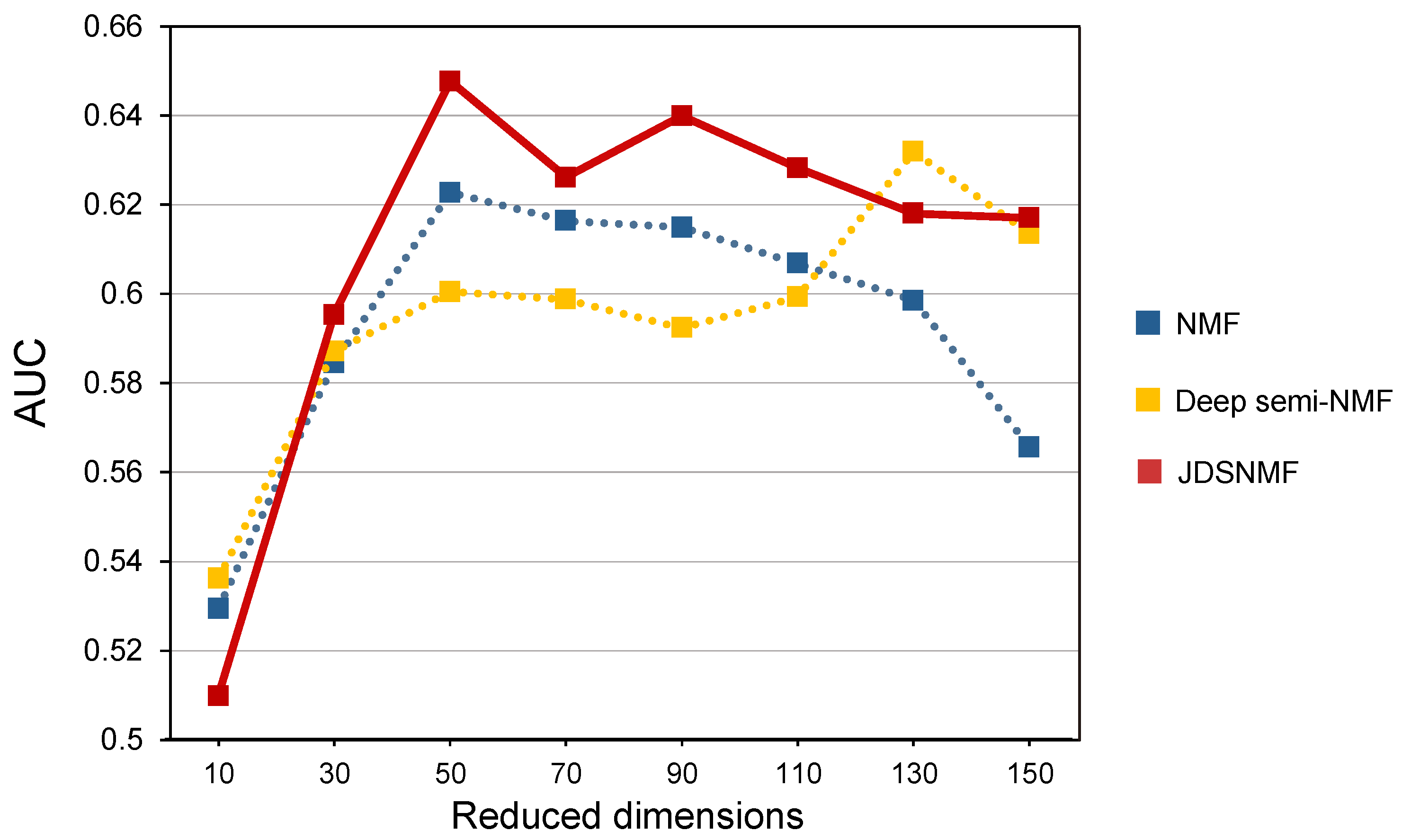
4.5. Classification Performance Comparison According to the Reduced Dimension K on ADNI Cohort
4.6. Module Analysis
4.6.1. AD-Related Module
4.6.2. Age-Related Module
4.7. Experiments on Bioimaging Data
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—A framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- De Vito, R.; Bellio, R.; Trippa, L.; Parmigiani, G. Multi-study factor analysis. Biometrics 2019, 75, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Žitnik, M.; Zupan, B. Data fusion by matrix factorization. IEEE Trans. Pattern Anal. Mach. Intell. 2014, 37, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, C.C.; Li, W.; Shen, H.; Laird, P.W.; Zhou, X.J. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic Acids Res. 2012, 40, 9379–9391. [Google Scholar] [CrossRef]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics 2015, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalise, P.; Fridley, B.L. Integrative clustering of multi-level ‘omic data based on non-negative matrix factorization algorithm. PLoS ONE 2017, 12, e0176278. [Google Scholar]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Lock, E.F.; Hoadley, K.A.; Marron, J.S.; Nobel, A.B. Joint and individual variation explained (JIVE) for integrated analysis of multiple data types. Ann. Appl. Stat. 2013, 7, 523. [Google Scholar] [CrossRef]
- Trigeorgis, G.; Bousmalis, K.; Zafeiriou, S.; Schuller, B.W. A Deep semi-NMF Model for Learning Hidden Representations. In Proceedings of the 31st International Conference on International Conference on Machine Learning—Volume 32 (ICML’14), Bejing, China, 22–24 June 2014; pp. II-1692–II-1700. [Google Scholar]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788. [Google Scholar] [CrossRef] [PubMed]
- Seung, D.; Lee, L. Algorithms for non-negative matrix factorization. Adv. Neural Inf. Process. Syst. 2001, 13, 556–562. [Google Scholar]
- Ding, C.H.Q.; Li, T.; Jordan, M.I. Convex and Semi-Nonnegative Matrix Factorizations. IEEE Trans. Pattern Anal. Mach. Intell. 2010, 32, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.M.; Tidor, B. Subsystem identification through dimensionality reduction of large-scale gene expression data. Genome Res. 2003, 13, 1706–1718. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; Jager, P.L.D.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Boutsidis, C.; Gallopoulos, E. SVD based initialization: A head start for nonnegative matrix factorization. Pattern Recognit. 2008, 41, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Sun, M.; Zhan, L.; Thompson, P.; Ji, S.; Zhou, J. Multi-Modality Disease Modeling via Collective Deep Matrix Factorization. In Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Halifax, NS, USA, 13–17 August 2017; ACM: New York, NY, USA, 2017; pp. 1155–1164. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. Tensorflow: A system for large-scale machine learning. In Proceedings of the 12th Symposium on Operating Systems Design and Implementation (16), Savannah, GA, USA, 2–4 November 2016; pp. 265–283. [Google Scholar]
- Xia, L.Y.; Wang, Y.W.; Meng, D.Y.; Yao, X.J.; Chai, H.; Liang, Y. Descriptor selection via log-sum regularization for the biological activities of chemical structure. Int. J. Mol. Sci. 2018, 19, 30. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin/Heidelberg, Germany, 2005; pp. 397–420. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why should i trust you?” Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, Y.; Mori, T.; Yoshida, T.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Funahashi, Y.; Iga, J.i.; Ueno, S.i. Elevated mRNA expression and low methylation of SNCA in Japanese Alzheimer’s disease subjects. J. Alzheimer’s Dis. 2016, 54, 1349–1357. [Google Scholar] [CrossRef]
- Li, R.; Yang, L.; Lindholm, K.; Konishi, Y.; Yue, X.; Hampel, H.; Zhang, D.; Shen, Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-β protein-induced neuron death. J. Neurosci. 2004, 24, 1760–1771. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Yang, L.; He, P.; Li, R.; Shen, Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J. Alzheimer’s Dis. 2010, 19, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Mol. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflamm. 2016, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Solerte, S.B.; Fioravanti, M.; Severgnini, S.; Locatelli, M.; Renzullo, M.; Pezza, N.; Cerutti, N.; Ferrari, E. Enhanced cytotoxic response of natural killer cells to lnterleukin-2 in alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1996, 7, 343–348. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Shegarfi, H.; Naddafi, F.; Mirshafiey, A. The role of natural killer cells in Alzheimer’s disease. Scand. J. Immunol. 2012, 76, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Butcher, S.; Killampali, V.; Lascelles, D.; Salmon, M. Neutrophil ageing and immunesenescence. Mech. Ageing Dev. 2001, 122, 1521–1535. [Google Scholar] [CrossRef]
- de Magalhaes, J.P.; Toussaint, O. GenAge: A genomic and proteomic network map of human ageing. FEBS Lett. 2004, 571, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, A.; Gal, Y.; Cipolla, R. Multi-task learning using uncertainty to weigh losses for scene geometry and semantics. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018; pp. 7482–7491. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathways | Overlap | q-Value |
|---|---|---|
| Ribosome | 23/153 | 7.35 × |
| Oxidative phosphorylation | 19/133 | 1.13 × |
| Parkinson’s disease | 18/142 | 1.75 × |
| Phagosome | 18/152 | 3.96 × |
| Influenza A | 19/171 | 3.46 × |
| Thermogenesis | 21/231 | 1.65 × |
| Viral myocarditis | 11/59 | 1.99 × |
| Graft-versus-host disease | 9/41 | 6.75 × |
| Non-alcoholic fatty liver disease (NAFLD) | 15/149 | 2.83 × |
| Leukocyte transendothelial migration | 13/112 | 2.73 × |
| Natural killer cell-mediated cytotoxicity | 14/131 | 2.61 × |
| Toxoplasmosis | 13/113 | 2.52 × |
| Tuberculosis | 16/179 | 4.18 × |
| Pathogenic Escherichia coli infection | 9/55 | 5.36 × |
| Leishmaniasis | 10/74 | 8.38 × |
| Alzheimer disease | 15/171 | 9.00 × |
| Tasks | NMF | Deep semi-NMF | JDSNMF | ||
|---|---|---|---|---|---|
| MRI | PET | MRI | PET | MRI, PET | |
| NL/MCI | 0.630 | 0.703 | 0.569 | 0.696 | 0.665 |
| MCI/AD | 0.831 | 0.811 | 0.838 | 0.826 | 0.843 |
| NL/AD | 0.935 | 0.963 | 0.949 | 0.972 | 0.973 |
| NL/SMC/EMCI /LMCI/AD | 0.624 | 0.677 | 0.678 | 0.717 | 0.718 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, S.; Lee, H. JDSNMF: Joint Deep Semi-Non-Negative Matrix Factorization for Learning Integrative Representation of Molecular Signals in Alzheimer’s Disease. J. Pers. Med. 2021, 11, 686. https://doi.org/10.3390/jpm11080686
Moon S, Lee H. JDSNMF: Joint Deep Semi-Non-Negative Matrix Factorization for Learning Integrative Representation of Molecular Signals in Alzheimer’s Disease. Journal of Personalized Medicine. 2021; 11(8):686. https://doi.org/10.3390/jpm11080686
Chicago/Turabian StyleMoon, Sehwan, and Hyunju Lee. 2021. "JDSNMF: Joint Deep Semi-Non-Negative Matrix Factorization for Learning Integrative Representation of Molecular Signals in Alzheimer’s Disease" Journal of Personalized Medicine 11, no. 8: 686. https://doi.org/10.3390/jpm11080686
APA StyleMoon, S., & Lee, H. (2021). JDSNMF: Joint Deep Semi-Non-Negative Matrix Factorization for Learning Integrative Representation of Molecular Signals in Alzheimer’s Disease. Journal of Personalized Medicine, 11(8), 686. https://doi.org/10.3390/jpm11080686