
Gastrointestinal Stromal Tumors—A Mini Review



 and
and 
Abstract
:1. Introduction
2. Classification of GIST
2.1. Etiological Classification of GISTs
- Carney–Stratakis syndrome (dyad).
- Carney triad syndrome.
- Family Neurofibromatosis type 1 (NF1).
2.2. Histological Classification of GISTs
- Spindle cell type (70%);
- Epithelioid type (20%);
- Mixed type (10%).
2.3. Immunohistochemical Classification of GISTs
2.4. Molecular Classification of GISTs
3. Clinical Manifestations of GISTs
4. Diagnostic Work-Up
- a.
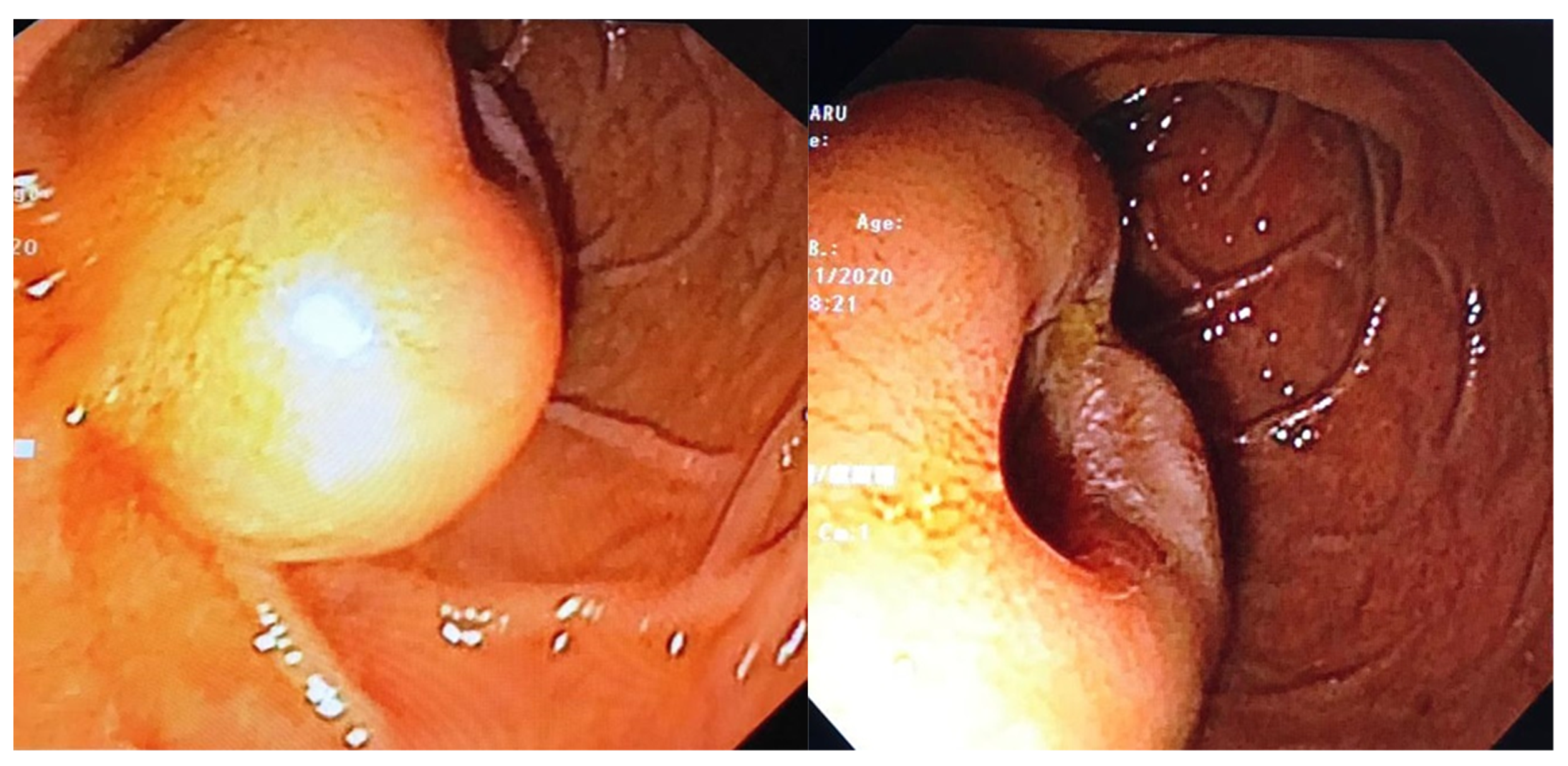
- Endoscopic examination has an essential role in definitive diagnosis because it allows the direct visualization of the tumor, with the possibility of biopsies for pathological examination (Figure 1). Both GISTs and leiomyomas may emerge as tumors with smooth margins located in the submucosa, with a normal mucosa cover that bulges into the lumen of the digestive tract. In some cases, a central ulceration may be seen.
- b.
- Endoscopic ultrasonography (EUS) permits the assessment of the invasion within the gastrointestinal wall and identification of the digestive tract layer as an origin for the GIST. Thus, most often, GISTs originate in the muscularis propria, and small lesions may also originate from the muscularis mucosa [40]. Upon EUS, GISTs appear as a hypoechoic, homogeneous tumor, with clearly defined edges, rarely irregular and sometimes with associated ulcers. EUS also enables both guided biopsies and GIST differentiation to other submucosal tumors [40].
- c.
- Contrast-enhanced computed tomography (CT) is the imaging method of choice to identify and describe the neoplasms, as well as to assess their extension and the presence of metastatic disease. Thus, CT allows the identification of metastases, which are most commonly located in the liver, omentum and peritoneal cavity. It also allows differential diagnosis, assessment of response to treatment and identification of tumor recurrence [41].
- d.
- Abdominal ultrasound, magnetic resonance imaging (MRI) and positron emission tomography (PET) are also useful in the evaluation of GISTs and in the detection of metastases. Although MRI has a diagnostic performance comparable to that of CT, CT scan remains the preferred initial imaging method used for staging the disease. There are some cases in which MRI may be a better imaging option, such as in GISTs found in specific locations (e.g., the rectum) or in evaluating the anatomical extension of surgery [42].
- e.
- The definitive diagnosis is histopathological. Biological samples may be obtained during endoscopic exploration, laparoscopic excision or laparotomy. In the case of metastases, the samples for histopathological diagnosis can also be obtained by biopsy of the metastases [43]. Depending on the tumor cell appearance after hematoxylin and eosin staining, three morphological types have been identified: spindle cell type, epithelioid type and mixed type [43,44]. Figure 2 shows the histological aspects of the spindle cell type in GISTs, and Figure 3 presents the histological aspects of the epithelioid type in GISTs.
- f.
5. Risk Stratification
- Very low risk: tumoral size <2 cm; mitotic count <5/50 high-power field (HPF).
- Low risk: tumoral size 2–5 cm; mitotic count <5/50 HPF.
- Intermediate risk: tumoral size <5 cm and mitotic count 6–10/50 HPF, or tumoral size 5–10 cm and mitotic count <5/50 HPF.
- High risk: tumoral size >5 cm and mitotic count >5/50 HPF, or tumoral size >10 cm and any mitotic rate, or any tumoral size and mitotic rate >10/50 HPF [10].
6. Treatment of GIST
- Type I: a tumor protruding into the digestive lumen while being narrowly connected to the muscularis propria.
- Type II: a tumor protruding into the digestive lumen while being widely connected to the muscularis propria.
- Type III: a tumor that is centrally localized on the gastric wall.
Despite All T
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors -definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows. Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, W.T.A.; Tielen, R.; Bonenkamp, J.J.; Lemmens, V.; Verhoeven, R.H.A.; de Wilt, J.H.W. Nationwide trends in the incidence and outcome of patients with gastrointestinal stromal tumour in the imatinib era. Br. J. Surg. 2018, 105, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.L.; Murphy, J.D.; Martinez, M.E.; Sicklick, J.K. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: Results of a population-based study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastrointestional Stromal Tumor—GIST: Statistics. Available online: https://www.cancer.net/cancer-types/gastrointestinal-stromal-tumor-gist/statistics (accessed on 30 June 2021).
- Kawanowa, K.; Sakuma, Y.; Sakurai, S.; Hishima, T.; Iwasaki, Y.; Saito, K.; Hosoya, Y.; Nakajima, T.; Funata, N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum. Pathol. 2006, 37, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Søreidea, K.; Sandvika, O.M.; Søreidea, J.A.; Vanja Giljacac, V.; Jureckovad, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Society of Clinical Oncology. Available online: https://www.cancer.net/ (accessed on 16 March 2021).
- Burch, J.; Ahmad, I. Gastrointestinal Stromal Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554541/ (accessed on 16 March 2021).
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Int. J. Surg. Pathol. 2002, 10, 81–89. [Google Scholar] [CrossRef]
- Yin, J.; Chen, J.D.Z. Roles of interstitial cells of Cajal in regulating gastrointestinal motility: In vitro versus in vivo studies. J. Cell Mol. Med. 2008, 12, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Blay, J.Y.; Hirota, S.; Kitagawa, Y.; Kang, Y.K. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer 2016, 19, 3–14. [Google Scholar] [CrossRef]
- Joensuu, H.; Hohenberger, P.; Corless, C.L. Gastrointestinal stromal tumour. Lancet 2013, 382, 973–983. [Google Scholar] [CrossRef]
- Graadt van Roggen, J.F.; van Velthuysen, M.L.; Hogendoorn, P.C. The histopathological differential diagnosis of gastrointestinal stromal tumours. J. Clin. Pathol. 2001, 54, 96–102. [Google Scholar] [CrossRef]
- Postow, M.A.; Robson, M.E. Inherited gastrointestinal stromal tumor syndromes: Mutations, clinical features, and therapeutic implications. Clin. Sarcoma Res. 2012, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Gopie, P.; Mei, L.; Faber, A.C.; Grossman, S.R.; Smith, S.C.; Boikos, S.A. Classification of gastrointestinal stromal tumor syndromes. Endocr. Relat. Cancer 2018, 25, 49–58. [Google Scholar] [CrossRef]
- Mussi, C.; Schildhaus, H.U.; Gronchi, A.; Wardelmann, E.; Hohenberger, P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin. Cancer Res. 2008, 14, 4550–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neppala, P.; Banerjee, S.; Fanta, P.T.; Yerba, M.; Porras, K.A.; Burgoyne, A.M.; Sicklick, J.K. Current Management of Succinate Dehydrogenase Deficient Gastrointestinal Stromal Tumors. Cancer Metastasis Rev. 2019, 38, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, H.; Hidaka, E.; Ota, H.; Minami, S.; Kajiyama, M.; Kuraishi, A.; Mori, H.; Matsuda, Y.; Wada, S.; Sodeyama, H.; et al. Familial gastrointestinal stromal tumor with hyperpigmentation: Association with a germline mutation of the c-kit gene. Gastroenterology 2001, 120, 210. [Google Scholar] [CrossRef]
- Hirota, S.; Nishida, T.; Isozaki, K.; Taniguchi, M.; Nishikawa, K.; Ohashi, A.; Takabayashi, A.; Obayashi, T.; Okuno, T.; Kinoshita, K.; et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology 2002, 122, 1493. [Google Scholar] [CrossRef]
- Fülöp, E.; Marcu, S.; Milutin, D.; Borda, A. Gastrointestinal stromal tumors: Review on morphology, diagnosis and management. Rom. J. Morphol. Embryol. 2009, 50, 319–326. [Google Scholar]
- Lasota, J.; Jasinski, M.; Sarlomo-Rikala, M.; Miettinen, M. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am. J. Pathol. 1999, 154, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Sobin, L.H.; Sarlomo-Rikala, M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod. Pathol. 2000, 13, 1134–1142. [Google Scholar] [CrossRef]
- Medeiros, F.; Corless, C.L.; Duensing, A.; Hornick, J.L.; Oliveira, A.M.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D. KIT-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. Am. J. Surg. Pathol. 2004, 28, 889. [Google Scholar] [CrossRef]
- Rubin, B.P.; Fletcher, J.A.; Fletcher, C.D. Molecular insights into the histogenesis and pathogenesis of gastrointestinal stromal tumors. Int. J. Surg. Pathol. 2000, 8, 5. [Google Scholar] [CrossRef]
- Loughrey, M.B.; Trivett, M.; Beshay, V.; Dobrovic, A.; Kovalenko, S.; Murray, W.; Lade, S.; Turner, H.; McArthur, G.A.; Zalcberg, J.; et al. KIT immunohistochemistry and mutation status in gastrointestinal stromal tumours (GISTs) evaluated for treatment with imatinib. Histopathology 2006, 49, 52–65. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865. [Google Scholar] [CrossRef]
- Lasota, J.; Corless, C.L.; Heinrich, M.C.; Debiec-Rychter, M.; Sciot, R.; Wardelmann, E.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Steigen, S.E.; Stachura, J.; et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod. Pathol. 2008, 21, 476. [Google Scholar] [CrossRef] [Green Version]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660. [Google Scholar] [CrossRef]
- Nannini, M.; Urbini, M.; Astolf, A.; Biasco, G.; Pantaleo, M.A. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J. Transl. Med. 2017, 15, 113. [Google Scholar] [CrossRef]
- Nannini, M.; Biasco, G.; Astolfi, A.; Pantaleo, M.A. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J. Med. Genet. 2013, 50, 653–661. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [Green Version]
- El-Menyar, A.; Mekkodathil, A.; Al-Thani, H. Diagnosis and management of gastrointestinal stromal tumors: An up-to-date literature review. J. Can. Res. 2017, 13, 889–900. [Google Scholar]
- Miettinen, M.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors of the stomach: A clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am. J. Surg. Pathol. 2005, 29, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Makhlouf, H.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors of the jejunum and ileum: A clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am. J. Surg. Pathol. 2006, 30, 477–489. [Google Scholar] [CrossRef] [PubMed]
- DeMatteo, R.P.; Lewis, J.J.; Leung, D.; Mudan, S.S.; Woodruff, J.M.; Brennan, M.F. Two hundred gastrointestinal stromal tumors: Recurrence patterns and prognostic factors for survival. Ann. Surg. 2000, 231, 51–58. [Google Scholar] [CrossRef]
- Watson, R.R.; Binmoeller, K.F.; Hamerski, C.M.; Shergill, A.K.; Shaw, R.E.; Jaffee, I.M.; Stewart, L.; Shah, J.N. Yield and performance characteristics of endoscopic ultrasound-guided fine needle aspiration for diagnosing upper GI tract stromal tumors. Dig. Dis. Sci. 2011, 56, 1757. [Google Scholar] [CrossRef]
- Bratu, O.G.; Cherciu, A.I.; Bumbu, A.; Lupu, S.; Marcu, D.R.; Ionita, R.F.; Manea, M.; Furam, C.; Diaconu, C.C.; Mischianu, D.L.D. Retroperitoneal tumors—Treatment and prognosis of tumor recurrence. Rev. Chim. 2019, 70, 191–194. [Google Scholar] [CrossRef]
- Scarpa, M.; Bertin, M.; Ruffolo, C.; Polese, L.; D’Amico, D.F.; Angriman, I. A systematic review on the clinical diagnosis of gastrointestinal stromal tumors. J. Surg. Oncol. 2008, 98, 384–392. [Google Scholar] [CrossRef]
- Gerrish, S.T.; Smith, J.W. Gastrointestinal stromal tumors—diagnosis and management: A brief review. Ochsner. J. 2008, 8, 197–204. [Google Scholar] [PubMed]
- Kapatia, G.; Gupta, N.; Saikia, U.N.; Gupta, P.; Rohilla, M.; Gupta, O.; Srinivasan, R.; Rajwanshi, A.; Dey, P. Fine needle aspiration cytology of primary and metastatic gastrointestinal stromal tumour. Cytopathology 2020, 31, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Pallag, A.; Rosca, E.; Tit, D.M.; Mutiu, G.; Bungau, S.G.; Pop, O.L. Monitoring the effects of treatment in colon cancer cells using immunohistochemical and histoenzymatic techniques. Rom. J. Morphol. Embriol. 2015, 56, 1103–1109. [Google Scholar]
- Miettinen, M.; Lasota, J. Histopathology of gastrointestinal stromal tumor. J. Surg. Oncol. 2011, 104, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.V.; Rigon, P.; Zettler, C.G.; Hartmann, A.A. Differential diagnosis of mesenchymal neoplasms of the digestive tract by cell block and immunohistochemistry. Cytopathology 2018, 29, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S. Differential diagnosis of gastrointestinal stromal tumor by histopathology and immunohistochemistry. Transl. Gastroenterol. Hepatol. 2018, 3, 27. [Google Scholar] [CrossRef]
- Bhatnagar, S.N. Gastrointestinal stromal tumor: Role of surgery and immunotherapy. J. Indian Assoc. Pediatr. Surg. 2010, 15, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Tirumani, S.H.; Baheti, A.D.; Tirumani, H.; O’Neill, A.; Jagannathan, J.P. Update on gastrointestinal stromal tumors for radiologists. Korean J. Radiol. 2017, 18, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef]
- Judson, I.; Bulusu, R.; Seddon, B.; Dangoor, A.; Wong, N.; Mudan, S. UK clinical practice guidelines for the management of gastrointestinal stromal tumours (GIST). Clin. Sarcoma Res. 2017, 7, 6. [Google Scholar] [CrossRef]
- Ahmed, M. Recent advances in the management of gastrointestinal stromal tumor. World J. Clin. Cases 2020, 8, 3142–3155. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, K.O. Management of gastric subepithelial tumors: The role of endoscopy. World J. Gastrointest. Endosc. 2016, 8, 418–424. [Google Scholar] [CrossRef]
- An, W.; Sun, P.B.; Gao, J.; Jiang, F.; Liu, F.; Chen, J.; Wang, D.; Li, Z.S.; Shi, X.G. Endoscopic submucosal dissection for gastric gastrointestinal stromal tumors: A retrospective cohort study. Surg. Endosc 2017, 31, 4522–4531. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, X.L.; Zhou, X.B.; Yang, H.; Zhu, L.H.; Chen, G.; Ye, L.P. Long-term outcomes of endoscopic resection for small (≤4.0 cm) gastric gastrointestinal stromal tumors originating from the muscularis propria layer. World J. Gastroenterol. 2018, 24, 3030–3037. [Google Scholar] [CrossRef]
- Astolfi, A.; Indio, V.; Nannini, M.; Saponara, M.; Schipani, A.; De Leo, A.; Altimari, A.; Vincenzi, B.; Comandini, D.; Grignani, G.; et al. Targeted Deep Sequencing Uncovers Cryptic KIT Mutations in KIT/PDGFRA/SDH/RAS-P Wild-Type GIST. Front. Oncol. 2020, 10, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann. Oncol. 2016, 27, 1794. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Reichardt, P.; Blay, J.-Y.; Boukovinas, I.; Brodowicz, T.; Broto, J.M.; Casali, P.G.; Decatris, M.; Eriksson, M.; Gelderblom, H.; Kosmidis, P.; et al. Adjuvant therapy in primary GIST: State-of-the-art. Ann. Oncol. 2012, 23, 2776–2781. [Google Scholar] [CrossRef]
- Joseph, C.P.; Abaricia, S.N.; Angelis, M.A.; Polson, K.; Jones, R.L.; Kang, Y.K.; Riedel, R.F.; Schöffski, P.; Serrano, C.; Trent, J.; et al. Optimal Avapritinib Treatment Strategies for Patients with Metastatic or Unresectable Gastrointestinal Stromal Tumors. Oncologist 2021, 26, e622. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935. [Google Scholar] [CrossRef]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Thomas, D.G.; von Mehren, M.; Patel, S.; Samuels, B.; Choy, E.; D’Amato, G.; Staddon, A.P.; Ganjoo, K.N.; et al. Association of Dasatinib With Progression-Free Survival Among Patients With Advanced Gastrointestinal Stromal Tumors Resistant to Imatinib. JAMA Oncol. 2018, 4, 814. [Google Scholar] [CrossRef] [PubMed]
- Nikfarjam, M.; Kimchi, E.; Shereef, S.; Gusani, N.J.; Jiang, Y.; Liang, J.; Sehmbey, M.; Staveley-O’Carroll, K.F. Surgical outcomes of patients with gastrointestinal stromal tumors in the era of targeted drug therapy. J. Gastrointest. Surg. 2008, 12, 2023–2031. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2018, 29 (Suppl. S4). [Google Scholar] [CrossRef]
- Koo, D.H.; Ryu, M.H.; Kim, K.M.; Yang, H.K.; Sawaki, A.; Hirota, S.; Zheng, J.; Zhang, B.; Tzen, C.Y.; Yeh, C.N.; et al. Asian Consensus Guidelines for the Diagnosis and Management of Gastrointestinal Stromal Tumor. Cancer Res. Treat. 2016, 48, 1155–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberini, J.L.; Al Nakib, M.; Wartski, M.; Gontier, E.; Cvitkovic, F.; Rixe, O.; Rougier, P.; Pecking, A.P. The role of PET scan in gastrointestinal stromal tumors. Gastroenterol. Clin. Biol. 2007, 31, 585–593. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, F.; Liu, S.; Guan, W. Comparative clinical features and short-term outcomes of gastric and small intestinal gastrointestinal stromal tumours: A retrospective study. Sci. Rep. 2019, 9, 10033. [Google Scholar] [CrossRef] [PubMed]
- Emory, T.S.; Sobin, L.H.; Lukes, L.; Lee, D.H.; O’Leary, T.J. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: Dependence on anatomic site. Am. J. Surg. Pathol. 1999, 23, 82–87. [Google Scholar] [CrossRef]
- Kiśluk, J.; Gryko, M.; Guzińska-Ustymowicz, K.; Kemona, A.; Kędra, B. Immunohistochemical diagnosis of gastrointestinal stromal tumors—An analysis of 80 cases from 2004 to 2010. Adv. Clin. Exp. Med. 2013, 22, 33–39. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Mutation | Characteristics |
|---|---|
|
|
| |
| |
| |
| |
| |
|
|
| |
|
|
| |
| |
|
|
| |
|
| Clinical Manifestations | Frequency |
|---|---|
| Overt or occult gastrointestinal bleeding | 28%—small intestine |
| 50%—stomach | |
| Incidental finding (asymptomatic) | 13–18% |
| Abdominal pain/discomfort | 8–17% |
| Acute abdomen | 2–14% |
| Asymptomatic abdominal mass | 5% |
| Mutational Status | TKI |
|---|---|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheorghe, G.; Bacalbasa, N.; Ceobanu, G.; Ilie, M.; Enache, V.; Constantinescu, G.; Bungau, S.; Diaconu, C.C. Gastrointestinal Stromal Tumors—A Mini Review. J. Pers. Med. 2021, 11, 694. https://doi.org/10.3390/jpm11080694
Gheorghe G, Bacalbasa N, Ceobanu G, Ilie M, Enache V, Constantinescu G, Bungau S, Diaconu CC. Gastrointestinal Stromal Tumors—A Mini Review. Journal of Personalized Medicine. 2021; 11(8):694. https://doi.org/10.3390/jpm11080694
Chicago/Turabian StyleGheorghe, Gina, Nicolae Bacalbasa, Gabriela Ceobanu, Madalina Ilie, Valentin Enache, Gabriel Constantinescu, Simona Bungau, and Camelia Cristina Diaconu. 2021. "Gastrointestinal Stromal Tumors—A Mini Review" Journal of Personalized Medicine 11, no. 8: 694. https://doi.org/10.3390/jpm11080694
APA StyleGheorghe, G., Bacalbasa, N., Ceobanu, G., Ilie, M., Enache, V., Constantinescu, G., Bungau, S., & Diaconu, C. C. (2021). Gastrointestinal Stromal Tumors—A Mini Review. Journal of Personalized Medicine, 11(8), 694. https://doi.org/10.3390/jpm11080694