Abstract
Tyrosine kinases inhibitors (TKIs) revolutionized chronic myeloid leukemia (CML) treatment for many years, prolonging patients’ life expectancy to be comparable to age-matched healthy individuals. According to the latest the European LeukemiaNet (ELN) recommendations, CML treatment aims to achieve long-term remission without treatment (TFR), which is feasible in more than 40% of patients. Nearly all molecular relapses occur during the first 6 months after TKI withdrawal and do not progress to clinical relapse. The mechanisms that are responsible for CML relapses remain unexplained. It is suggested that maintaining TFR is not directly related to the total disposing of the gene transcript BCR-ABL1, but it might be a result of the restoration of the immune surveillance in CML. The importance of the involvement of immunocompetent cells in the period of TKI withdrawal is also emphasized by the presence of specific symptoms in some patients with “withdrawal syndrome”. The goal of this review is to analyze data from studies regarding TFRs in order to characterize the elements of the immune system of patients that might prevent CML molecular relapse. The role of modern droplet digital polymerase chain reaction (ddPCR) and next-generation sequencing (NGS) in better identification of low levels of BCR-ABL1 transcripts was also taken into consideration for refining the eligibility criteria to stop TKI therapy.
1. Introduction
Chronic myeloid leukemia (CML) is the first malignant monoclonal disease of hematopoietic stem cells (HSCs) with identification of an acquired chromosomal abnormality: the Philadelphia chromosome (Ph) [1]. This chromosome 22 is the result of a balanced exchange of genetic material between the long arms of chromosome 9 and 22 (translocation, t(9;22)(q34;q11)), generating juxtaposition of the BCR gene in 9q to the ABL1 proto-oncogene in 22q [2,3]. Expression of this fusion gene produces the BCR-ABL1 oncoprotein whose tyrosine kinase activity deregulates many signal transduction pathways in the HSC. The BCR-ABL tyrosine kinase activity represents the primary alteration in CML, proved in vitro as well as in animal models. The oncogenic BCR-ABL1 protein (P210BCRABL1) is responsible for the phosphorylation of tyrosine residues on individual substrates, such as adapter, catalytic, cytoskeleton, and membrane proteins. Moreover, due to autophosphorylation, binding sites are created for the SH2 domains of other proteins by increasing phosphotyrosine on BCR-ABL1. Malignant transformation mainly concerns mechanisms related to altered adhesive properties, activation of mitogenic signaling pathways, inhibition of apoptosis, and degradation of inhibitory proteins. The inhibition of the adhesion of CML progenitor cells to the bone marrow stromal cells and the extracellular matrix is mainly related to the influence of BCR-ABL1 on integrin dysfunction. Mitogenic signaling is important for the pathogenesis of Ph-positive cells. BCR-ABL1 protein has been shown to be associated with disturbances in Ras mitogen-activated protein kinase (MAPK), leading to increased proliferation. The Jak-Stat pathway, and more precisely, constitutive phosphorylation of the transcription factors Stat1 and Stat5, lead to the impairment of transcriptional activity and promote the anti-apoptotic effect. The phosphoinositide 3-kinase (PI3K) induces the proliferation of leukemic cells, thereby inhibiting apoptosis, which may also be affected by Myc overexpression. Maliciously transformed BCR-AB1 cells prevent apoptosis by blocking the activation of caspases and thanks to the aforementioned Ras protein or PI3 kinase [4]. The natural evolution of CML consists of three phases: a chronic phase (CP-CML) that without tyrosine kinases inhibitor (TKI) therapy could last 3–4 years with harmonic malignant HSC differentiation; an accelerated phase (AP-CML) with HSC maturation hiatus and a blastic phase (BP-CML) in acute leukemia—myeloblastic or lymphoblastic—with a fatal outcome within a few months. This three-phase evolution has been modified by the clinical results of TKIs, and since then CML is no longer a fatal disease but rather a chronic condition with age-related diseases. TKIs revolutionized CML treatment for many years, and prolong patients’ life expectancy to be comparable to age-matched healthy individuals [5,6,7,8,9]. With significant advances in the treatment of CML, priority is placed on improving the quality of life of CML patients and on the efficacy, tolerability, and toxicity of TKIs, as well as on the pharmacoeconomics of long-term treatment. Therefore, the ability to discontinue TKI treatment and achieve long-term remission without treatment (treatment-free remission—TFR) has become a new goal in CML, and the selection of appropriate therapy is a key element in the depth and speed of achieving a very deep molecular response.
2. Treatment
Symptomatic patients with leukocytosis and thrombocytosis may receive a dose of hydroxyurea before confirming a diagnosis of CML for a short time. Pregnancy is an absolute contraindication to the treatment of TKI, then interferon alpha (IFN-α) therapy is recommended. Currently, the advent of pegylated interferon (PEG-IFN) has appeared, which is more effective and better tolerated. Recent studies have shown that PEG-IFNα in combination with imatinib can accelerate the speed and depth of molecular responses [10], but this requires further research similarly to the combinations with the second-generation (2G) TKIs with PEG-IFNα, which could also generate benefits [11,12]. Five drugs are approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of CML: imatinib; second-generation TKIs, nilotinib, dasatinib, bosutinib; and third-generation TKI, ponatinib. Rodotinib, another 2G TKI, has only been approved in South Korea. Generic imatinibs are alternative drugs for the first-line treatment, which are already widely available and became affordable with significantly lower costs than the brand product [13,14,15,16].
The general principle of operation of most used TKIs is to inhibit the activity of ABL1 tyrosine kinase in an ATP-competitive manner with different specificities. Treatment of BCR-ABL1 transformed cells inhibits their proliferation and induces apoptosis. However, it has been proven that despite maintaining a stable deep molecular response (DMR), leukemic cells can still be detectable but might be under the control of the immune system [17]. Imatinib binds to the ABL1 kinase domain when it has a catalytically inactive conformation. The mutations in the binding site of imatinib (within the SH2 contact, P loop positions: M244, G250, Q252, Y253, E255, T315, F317, M351, F359, H396) cause resistance to this drug, e.g., promoting the active conformation of the enzyme [18]. Nilotinib, despite its increased affinity, binds to the same pocket as imatinib, therefore therapy with this drug is also associated decreased activity against mutations in positions: Y253, E255, T315, F359, but it is effective against some mutations resistant to imatinib [19]. In parallel with the advancement of nilotinib, a dual SRC/ABL inhibitor dasatinib was developed, which binds to the active conformation of the ABL1 kinase, showing even higher affinity than previous drugs. The dasatinib resistance mutations are V299, T315, and F317 [20]. A similar binding mechanism to dasatinib is demonstrated by bosutinib, the other SRC/ABL inhibitor with mutations resistant to this drug similar to that of dasatinib, and additionally G250 and E255. Table 1 summarizes the first- and second-generation TKIs in the treatment of CML.

Table 1.
First- and second-line-generation TKIs.
Ponatinib is 3G TKI drug, potent inhibitor of ABL1 kinase, and is effective against the described resistance mutations to other TKIs, including T315I. The T315I mutation, which is a point mutation, and due to its location at the entrance to the ATP binding site determines resistance to imatinib, nilotinib, dasatinib, and bosutinib. In different trials, ponatinib showed inhibitory activity against native BCR-ABL1 kinase and several ABL1 mutations. For this reason, ponatinib is currently indicated for the treatment of CML in every phase of the disease. The risk of resistance to ponatinib may be due to several general drug-related mechanisms, such as inhibition of single or multiple non-therapeutic target kinases, possible drug–drug interactions, or direct pharmacological toxicity, but may be due to the occurrence of still unknown mutations. However, due to the broad target profile of ponatinib, it has a high risk of vascular occlusive events, which has limited its potential in the treatment of CML in the first line but represents the drug of choice for T315I mutants as well as in subsequent therapy lines [30]. The T315I point mutation has long been a clinical challenge, so a new promising drug, asciminib, which was first approved by the FDA in February 2021, is an attractive approach to the treatment of CML, especially for patients with the T315I mutation. Asciminib is a STAMP (specifically targeting the ABL myristoyl pocket) inhibitor that does not bind to the ATP binding pocket and thus exhibits a mechanism of action different from that of commonly used TKIs. Asciminib as an allosteric ABL1 kinase inhibitor stabilizes the inactive conformation of the enzyme by binding to the ABL1 myristoyl pocket. The encouraging results of asciminib have shown its safe and clinically effective profile and, moreover, it might be a therapeutic option in patients with T315I, including those with ponatinib resistance/intolerance [31]. Another new substance—olverembatinib, known as HQP1351—also proved effective in the II phase of clinical trials in CML patients with a broad spectrum of mutations, including the T315I mutation [32]. Vodobatinib was studied in a phase I clinical trial in a group of CML patients who failed more than three different TKIs divided into groups not treated with ponatinib as well as those treated. Thanks to the comparative effectiveness in both groups, it may become a promising agent for the treatment of resistant CML [33]. When resistance occurs on the first-line drug, it is obligatorily changed to another TKI. Each change to a different TKI is undertaken by the existing mutations, age, comorbidities, and individual preferences of the patient [34]. In particular, the risk of cardiovascular complications should be taken into account when ponatinib is started [35,36]. The PACE study showed that ponatinib provided sustained and clinically meaningful responses that were sustained even after dose reduction [37], but the incidence of arterial occlusive events (AOEs) is relatively high [35].
The most mature data for TFR and the availability of the generic version supports the primary utilization of imatinib for initial TFR attempts. In fact, the use of 2G TKIs as first-line treatment leads to faster molecular responses [38] and patients treated with nilotinib or dasatinib in the first line develop fewer resistance mutations to other TKIs and therefore can be eligible for TFR faster, as demonstrated in the DASSION study [21] and ENESTnd [22]. However, there are some concerns regarding the toxicity of 2G TKIs, which are more toxic then imatinib. In more than 50% of medium- and high-risk patients, as assessed by the new EUTOS long-term survival (ELTS) score, the benefits of 2G TKIs were reported in terms of extended overall survival (OS). The BFORE study showed that bosutinib used as subsequent line treatment in patients with resistance or intolerance to prior therapies was more effective at generating faster and deeper molecular responses than imatinib, but more frequent adverse events, such as diarrhea and elevated levels of aminotransferases, were reported [28]. Furthermore, 2G TKIs are also very effective in patients diagnosed with CML in the accelerated phase. A French study reported that after 7 years of follow-up, the OS in these patients was 87% and the PFS was 83%.
3. AlloSCT
Until now, the only potential cure for CML has been allogeneic stem cell transplantation (alloSCT). However, nowadays, alloSCT is a suitable option only for a limited group of patients due to the effectiveness of TKIs. The lack of a sustained response to 2G TKIs and ponatinib suggests a high risk of disease progression, which is an indication for an alloSCT. Patients with AP-CML or progressing to AP-CML also qualify for alloSCT due to the high risk of disease progression. In BP-CML, alloSCT is not recommended, thus every effort should be made to achieve a second CP-CML and then perform an alloSCT immediately [39,40,41].
4. Monitoring and Prognostic Factors
Due to the detection of additional aberrations, classical cytogenetics is of importance, especially at the time of diagnosis. Cytogenetics testing should be performed in patients with atypical translocations or BCR-ABL1 transcripts, treatment resistance to exclude additional chromosomal aberrations (ACA), and with progression to AP-CML/BP-CML. The FISH method may also be helpful in detecting abnormal transcripts. Although it is a good quantitative tool, it is currently not recommended for use when considering TFR [42].
The molecular response (MR) is the ratio of BCR-ABL1 transcripts to ABL1 transcripts or to other control transcripts according to the international scale (IS) and is expressed as BCR-ABL1% on a log scale according to the IRIS study (BCR-ABL1 1% correspond to a decrease of 2 logs, respectively) [43,44]. The goal for the first year of therapy is to achieve major molecular remission (MMR). Soverini et al. emphasize changing the common control gene ABL1 to GUSB in the first months of treatment, due to the underestimation of the actual BCR-ABL1/ABL1 ratio in the initial stages of therapy, which is crucial. This is due to the fact that the PCR primers used for ABL1 amplification also amplify the target sequence from the BCR-ABL1 fusion transcript, while the use of GUSB as a control gene or performing a parallel assessment of ABL1 and GUSB could improve the reliability of the results [18,45,46].
The Spanish TKI discontinuation report confirms the existence of two prognostic factors: length of TKI treatment, which should be at least 5 years (TFR: 59% with MR4.5 > 5 years vs. 30% with MR4.5 < 5 years), and duration of deep molecular response (MR4.5) for at least 4 years (TFR: 47% with MR4.5 > 4 years vs. 25% with MR4.5 < 4 years).
So far, to determine the prognosis of the disease in CML patients, there have been three systems—Sokal, Euro, and EUTOS. However, the ELTS score was created, which captures the same prognostic factors as the Sokal score, such as basic morphological data, spleen size, and age, but the age value has a lower negative prognostic value than the Sokal score and is important in predicting the likelihood of death related to leukemia [47,48].
The latest recommendations the European LeukemiaNet (ELN) prefer the use of ELTS because it delimits the risk groups in patients much better than the Sokal score and about 50% of patients can change allocations based on the ELTS score [49]. In the group of patients over 65 years of age, it turned out that the ELTS score identifies a group of patients who have a greater chance of achieving MMR or MR4. It is recommended for risk assessment in this group of CML patients, where it may be important in deciding to start 2G TKIs treatment as soon as possible, and even from the very beginning. Additionally, it identifies a group of patients who do not require such treatment, especially when patients are at risk of additional age-related comorbidities.
5. The Emerging Role of New Methods of Molecular Testing in CML-NGS and ddPCR
Next-generation sequencing (NGS) and digital droplet PCR (ddPCR) are molecular techniques that are increasingly being considered in terms of mutation detection in CML patients. Sanger sequencing (SS) is still the gold standard in screening tests for the detection of BCR-ABL1 kinase domain (KD) mutations, but recent studies on NGS have shown its importance especially in the detection of low-level mutations. The NEXT-in-CML study showed that mutations undetectable by SS occurred in 34% of patients and 18% (of all) had low-level mutations [50]. Due to the kinetics of TKI, resistant low-level mutations invariably expand if the patients are not switched to another TKI or appropriate TKI dose, which may be important in clinical decision-making and thus NGS should enter clinical practice. Despite the effectiveness, NGS remains an expensive and technically demanding method. In recent years ddPCR has been gaining popularity due to the linearity of the results, their accuracy, repeatability, and standardization. The greatest advantage of ddPCR is a sensitivity of around 107, which is definitely beyond the limits of RQPCR [42]. A good correlation was observed between the results of ddPCR and NGS in detecting resistance mutations [51].
6. Treatment-Free Remission (TFR)
The clinical course of CML has changed significantly in recent years [52]. CML is identified with a chronic disease in which the effectiveness of targeted therapy translates into a much better prognosis of patients who live like healthy people [5,8,9]. Moreover, patients with CML are dying not directly as a consequence of this disease, but due to age-related comorbidities [49]. The concept of an “operational cure” emerged in connection with the increasing questions about improving the quality of life of CML patients, preventing the toxicity of long-term drug intake, and alleviating their side effects. It was based on keeping the patient in remission despite the detection of leukemic cells in minimal residual disease, which may be related to the immunological control and the type of BCR-ABL1 transcript [53,54]. Discontinuation of TKI treatment and the achievement of sustained TFR is possible in patients who have achieved a stable deep molecular response (DMR). A DMR is understood as BCR-ABL1 transcript levels of molecular response MR 4, MR 4.5, and MR5 on the IS (Table 2) [44,55].
Table 2.
Scoring molecular response.
In the consideration of the latest recommendations of ELN of March 2020, the aim of CML treatment is “normal survival and good quality of life without life-long treatment” [49]. Consequently, it is important to standardize the recommendations and criteria for qualifying patients for an effective TFR. In the new approach, it is possible to maintain a stable DMR without continuing treatment with TKI (Figure 1). Over time, successive attempts have been made to maintain TFR, but most are still limited to clinical trials [56].
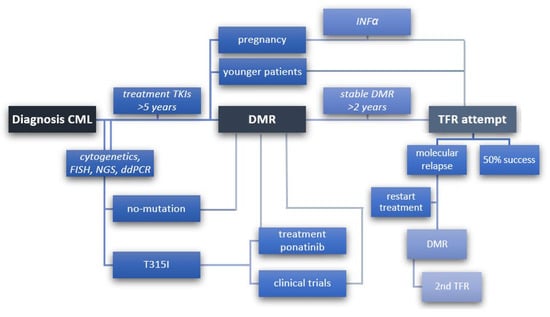
Figure 1.
Steps to follow to achieve TFR from diagnosis of CML, detection of individual mutations, treatment of TKI to stable deep molecular remission (DMR), and then discontinuation of treatment (TFR) with 50% success or molecular relapse, prompting re-initiation of treatment to re-attain DMR and eventually make a second attempt to discontinuation treatment (2nd TFR). A scheme to include younger patients with low- to medium-risk disease and women who wish to become pregnant for whom TFR has a high priority.
An important issue in favor of undertaking TFR studies is the toxicity of TKIs use, which may last for many years due to the prolonged exposure of the drug to the organism. The reported adverse events may be hematological, including neutropenia, thrombocytopenia, and anemia. They are not the basis for changing the treatment and very rarely cause complications, as often the stabilization of hematological parameters requires time for the organism to adjust to the drug. Non-hematological adverse events are those that worsen the patient’s quality of life and complications that require a change in treatment due to the risk of deterioration of health or even death [49].
The implementation of TFR is justified, especially in patients who wish to become pregnant. Pregnancy in patients with CML precludes continuing TKI treatment due to their teratogenic effect [57]. It is probably related to the inhibition of organogenesis by blocking the PDGFR receptor by TKIs [49]. Women who cannot undergo TFR due to a lack of a sustained DMR can replace TKIs with IFN-α therapy.
Children are another group, apart from pregnant women, for whom a successful TFR might be of great prominence. Although the CML is rarely diagnosed in children, it is speculated that treatment may have a negative impact on their growth [58,59,60]. Currently, TFR is not attempted in pediatric patients because the clinical course of the disease is usually more aggressive than in adults [61]. However, studies in this group of patients are necessary due to exposure to the long-lasting toxicity of TKIs according to the principle of lifelong treatment.
The prevalence CML increases with better survival rates of CML patients and more than 400,000 CML patients are expected in Europe by 2050 [56,62]. The cost of treating one patient is currently EUR 30–40,000 per year in most European countries, and although these costs are expected to fall due to the availability of generic imatinib and soon also generic dasatinib, it is still a great impact on the healthcare budget [15,63].
Table 3 summarizes the results of TFR trials. TFR is in the range of 30–70%, and its rate decreases with time after treatment discontinuation in subsequent attempts. Molecular relapse has been defined in most studies as a loss of MMR, followed by treatment resumption [64]. Overall, the median molecular relapses based on the above studies is 47%, with a similar relapse rate in the discontinuation studies with both imatinib and nilotinib or dasatinib. Thus, the probability of maintaining TFR seems to be similar for both imatinib and 2G TKIs nilotinib or dasatinib discontinuation and is approximately 50%. Eighty percent of all molecular relapses occur within the first 6–8 months after TKI discontinuation [65]. TFR studies do not envisage the participation of patients treated with TKIs 3G, but such studies have been conducted. Six patients from the PAECE study entered the TFR trial, four of which are still in remission, and two experienced molecular relapse but regained DMR after re-initiation of low-dose ponatinib treatment [66].
Table 3.
Clinical trials of TFR.
The second stop attempt has a significantly lower rate of successful TFR. The median molecular relapse in the group of patients undergoing 2nd TFR in the above studies reached 74%. Second attempts to stop TKI in MR4.5 were successful in only 1/3 of eligible patients. The latest reports confirmed that TFR2 trials are safe and that discontinuation of treatment may be effective, but require close monitoring of the patient in an extended stage. Decisive for the success of TFR2 is the duration of the TFR for the first attempt and thus the rate of molecular relapse and the TKI-free time after the first attempt. It seems that the MR depth should be at the minimum level of MR4.5 and its duration is at least 2 years for successful TFR2.
Most studies used sustained DMR at MR4 or MR4.5 levels as eligibility criteria for more than a year or two. The duration of TKI treatment at the time of patient recruitment was also important for eligibility, which was on average around 2–3 years. The largest TFR study—the EURO-SKI—has shown that a longer duration of TKI and DMR treatment, as well as prior IFN-α therapy, correlate with a higher percentage of effective TFR [90]. Furthermore, the longer duration of TKI treatment relates to a DMR. It has been reported that the success of TFR is significantly lower in patients with stable MMR who have not achieved DMR [91]; however, this requires further research. Upon re-initiation of therapy, 90–95% of patients regain their initial molecular level or at least regain MMR. The ELN expert panel highlights that some patients have fluctuations in BCR-ABL1 level, which sometimes improve over time without restarting therapy [49]. Although careful comparison of TFR research studies is limited by differences in eligibility criteria and definitions of DMR as well as molecular relapse, these have established standards for the safe and effective conduct of TFR studies (Table 4).
Table 4.
Requirements for tyrosine kinase inhibitor discontinuation.
The recent ELN recommendations have underlined the importance of long-term systematic monitoring of patients after treatment is stopped due to the possibility of late relapses.
Patients’ motivation and attitude towards TFR seem to be very important, especially for women wishing to become pregnant and younger patients. Additionally, in such patients, the option of switching to 2G TKI therapy may be considered for achieving a faster and deeper molecular response, provided the DMR has been achieved so far. However, there are also patients who do not want to discontinue treatment despite meeting the inclusion criteria and such patients should not be included in the TFR trials.
7. Immune System-Specific Markers in CML
The clinical course of CML involves not only the dysregulation of tyrosine kinase function, but also manifests as a deep dysfunctional immune response against tumor cells expressing the fusion gene BCR-ABL1. In CML, like in other malignancies, the immune response against cancer is impaired, resulting in immune escape of the malignant cells, which supports cancer development. Active immunotherapy represents a tempting approach directing not only the immune response against leukemic cells but also restoring immune function. Searching for the most meaningful immunogenic leukemia-associated antigens as novel targets for immunotherapy and assessment of CML-specific immune response in CML patients could help to characterize the role of TKIs in promoting a long-term response. Recent studies proved the efficient induction of specific cytotoxic T lymphocytes (CTLs) directed against leukemia-associated antigens: BCR-ABL1, the receptor for hyaluronan-mediated motility (RHAMM), Wilms Tumor-1 (WT-1), proteinase 3 (PR3), preferentially expressed antigen in melanoma (PRAME), M-phase phosphoprotein (MPP11), Aurora A kinase (AURKA), and human chromosome X open reading frame 48 (CXorf48) in CML [92,93,94,95,96,97,98,99,100]. Other antigens like renal cell carcinoma-associated antigen (NEWREN60) and sperm-associated antigen 9 (SPAG9) demonstrated restricted and upregulated mRNA expression in CML, although they have not been functionally investigated yet [92,101,102]. Moreover, patients with CXorf48-specific CTL had a higher rate of successful TFR after imatinib than those without [103]. All of these molecules represent novel promising targets for peptide-based immunotherapy and they could act as markers for assessment immune responses in CML patients with a deep molecular response.
Regulatory T cells (Treg) are a group of immunosuppressive T cells that play a crucial role in self-tolerance and immune response against tumor cells. Characteristic features of Tregs cells are their anergic state, the ability of active inhibition of CD4+CD25- T cells, CD8+ T cells, dendritic cells (DC), natural killer (NK) cells, natural killer T (NKT) cells, and B cells in a cell-to-cell contact and dose-dependent manner. It may facilitate tumor cell proliferation and it is one of the mechanisms of tumor cells’ escape from immune surveillance. Increased proportions or functional activities of Treg are found in patients with solid or hematological malignancies, thereby providing strong evidence that Treg may play a critical role in suppressing an effective antitumor immune response [104]. Bachy et al. [105] have shown that Treg are significantly increased in CML patients with intermediate- or high-risk Sokal scores when compared with low-risk patients. Furthermore, Zahran et al. [106] found an increased percentage of Tregs in CML patients in comparison to healthy controls. They also found that Treg numbers were lower in patients with chronic phase CML versus accelerated and blast phases and were significantly decreased in patients with CMR when compared to those patients without CMR. Similar reports have been given by Hus et al. [107], who reported that the percentage of CD4+CD25highFoxP3+ cells was higher in untreated CML patients compared to healthy controls. Additionally, Rojas et al. [108] demonstrated an increased frequency of Treg in patients with high levels of BCR-ABL1 transcripts than in cases with low levels of BCR-ABL1 gene transcripts or healthy donors. In another study, Nadal et al. [109] observed that Tregs cells are increased in patients after alloSCT when compared with healthy controls (median 1.5 vs. 0.78%, p = 0.004). Interestingly, the analysis of the newly diagnosed CML patients showed that not only the Tregs frequency was markedly reduced compared to patients after alloSCT (median 0.27 vs. 1.5%, p = 0.0003), but also with respect to healthy volunteers (median 0.27 vs. 0.87%, p = 0.03). Moreover, they found significantly higher Treg cells numbers in CML patients that relapse after alloSCT, suggesting that these cells may be detrimental to the graft vs. leukemia effect of allogeneic stem cell transplantation. Several studies have reported a complex influence of TKI, such as imatinib, on the improvement of the immune system function. Such therapy on the one hand has a positive effect by enhancing anti-tumor function of CD4+ T cells and dendritic cells, but on the other hand might reduce the proliferation and activity of Treg cells. Data from in vitro and in vivo studies on mice models reported a significant downregulation of proliferation, activation, FOXP3 expression, and an impaired immunosuppressive function of CD4+CD25high Tregs by imatinib in a dose-dependent manner [110]. Lu et al. [111] showed that treatment with TKI reduces the percentage of T, Treg, CD4+, and CD8+ T cells to a different extent depending on the TKI used in CML patients compared to healthy controls. The decrease in the amount of Treg gradually deepened with the duration of the treatment. This study also demonstrated inhibition of Treg function, including proliferation, suppression, and expression of IL-4, IL-10, TGF-β cytokines, and FOXP3, GITR, and CTLA-4 molecules in the imatinib and dasatinib treatment groups, which was not observed in patients treated with nilotinib. A study by Dai et al. [112] indicated that in CML patients treated with dasatinib, it has the strongest effect on increasing the Th1 percentage compared to imatinib and nilotinib treatment, while reducing the Treg lymphocyte population, thus increasing the chance of successfully achieving MMR and MR4.5. Important observations were made by Alves et al. [113], who proved that CML patients treated with 2G TKI had a lower percentage of CD4+ Treg and granulocytic myeloid-derived suppressor cells (Gr-MDSC) compared to patients receiving imatinib (median CD4+ Treg 3.63 vs. 6.18%, p = 0.005 and Gr-MDSC 4.2 vs. 8.2%, p = 0.003), but higher levels of PD-1-co-expressing CD4+ cells (1.92 vs. 1.0%; p = 0.001).
CML patients with low levels of CD62L on T lymphocytes and high levels of soluble CD62L in plasma induce a pro-inflammatory leukemic environment, while nilotinib increases CD62L expression on T lymphocytes and thus improves antitumor immunity [114]. In turn, dasatinib significantly supports the anti-leukemic response by inducing the expansion of large granular lymphocytes (LGL), such as NK cells and CD8+ T cells. Due to the generation of a strong cytotoxic memory response, LGLs can effectively destroy cancer cells even after cessation of treatment [115,116].
Another subpopulation of the blood cells that might play a crucial role in immune system restoration after TKI treatment is natural killer (NK) cells. NK cells are effector lymphocytes of the innate immune system that control several types of tumors and microbial infections by limiting their spread and subsequent tissue damage. They are defined as CD3−CD56+ lymphocytes, distinguished as CD56bright and CD56dim subsets. Activated NK cells are in a position to directly or indirectly exert their antitumor activity to control tumor growth and prevent rapid dissemination of metastatic tumors by immune surveillance mechanisms [117]. The inhibitory killer cell immunoglobulin-like receptors (KIR) and their human leukocyte antigen (HLA) ligands play a crucial role in the antitumor activity of NK cells, and their mismatch in the context of the major histocompatibility complex (MHC) may trigger alloreactivity of NK cells [118]. Moreover, NK cell alloreactivity is of great importance in controlling a graft-versus-host (GVH). Interestingly, the genotype of the KIR receptor is also important as the presence of KIR3DS1/KIR3DL1/HLA-Bw4 was shown to be significantly associated with relapse after the TFR test. Cumulative TFR is significantly higher in patients homozygous for the KIR A haplotype. Previously reported results indicate that NK cells of newly diagnosed CML patients are in reduced number or proportion among lymphocytes and have limited cytolytic capacity at the diagnosis of CML [99,119,120]. A key role in the cytotoxicity of NK cells is played by receptors activating mainly NKG2-D type II integral membrane protein (NKG2D) and their NKG2DL ligands, including MICA, MICB, ULBP1, and ULBP2, which are expressed on CML blasts. Although there is a lack of specific prognostic factors that could determine which patients could discontinue the therapy, there is increasing evidence suggesting that NK cells are important in the control of leukemic growth. In animal experiments, it has been shown that NK cells after implanting them in the irradiated bone marrow of the recipient mice can control leukemic cells [121]. In CML patients, increased NK cell counts seem to correlate with successful imatinib discontinuation. Boissel et al. [122] demonstrated that at the time of diagnosis, patients with CML had abnormally high levels of soluble forms of MIC (sMIC) in the serum and poor NKG2D expression on NK and CD8+ T lymphocytes, which were restored by imatinib therapy. Ohyashiki et al. [123] reported that CML patients who sustain a CMR after imatinib discontinuation have higher levels of NK cells than normal subjects or CMR patients under imatinib therapy do. In other study, Mizoguchi et al. [124] observed a significantly higher percentage of effector populations of NK cells after stopping imatinib in CMR groups than in the fluctuating CMR and control groups. The elevated levels of these effector NK cells were sustained for more than 3 years after imatinib discontinuation. In addition, imatinib treatment activates NK to the intensified release of IFN-γ in gastrointestinal stromal tumor patients [125]. The most recent studies reported that after imatinib discontinuation, the NK cell level significantly increased in non-relapsing CML patients and remained higher in comparison with relapsing patients [126]. Moreover, Ilander et al. [127] suggested that non-relapse patients demonstrated a higher than the median CD56dim NK cell subset percentage at the time of imatinib withdrawal and had a better probability of staying in remission. NK cells have been an attractive tool for several years in cancer immunotherapy, an important approach of which is to block checkpoints, such as interactions programmed death-1 (PD-1)/programmed death ligand-1 (PDL-1), IL-1, and JAK/STAT pathways [120].
These results suggest that the immunological activation status of NK cells contributes to DMR maintenance. Higher activation levels of effector NK cells in CML patients treated with TKI might reflect minimization of BCR-ABL1 transcript levels and therefore could serve as additive information when determining imatinib discontinuation. The function and genetic determinants of the NK-cell expansion need further study, but it is tempting to speculate that in the future, successful CML therapy should aim to increase the number and activity of NK cells. Recently, an additional NK subpopulation of interesting T-cell properties (NKT) that might play a role in the anti-leukemia immune response was defined. Invariant NKT cells (iNKT) have been reported to be dysfunctional, but their function improved in patients who achieved CCyR with IFN-α or TKI treatment [128]. There is no data about the function of NKT cells in patients who stopped TKI therapy.
TKIs affect a broad spectrum of kinases, thereby exhibiting a negative impact on the proliferation and activation of multiple different blood cell types, including T cells [129]. Recent data suggest that inhibition of dendritic cell (DC) generation in imatinib-treated patients contributes to a reduction of CTL levels, due to ineffective peptide priming. Moreover, imatinib treatment seems to directly inhibit T cell receptor-mediated proliferation and activation results in delayed-type hypersensitivity in CML patients. In turn, long-term imatinib treatment might also induce the immune response of CD4+ T cells with the generation of CD4+ and CD8+ BCR-ABL1 specific effector-memory T cells [130,131]. All of these studies emphasized the important role of imatinib in the modulation of anti-leukemic response in CML patients. Plasmacytoid dendritic cells (pDCs) can induce immunosuppressive Treg lymphocytes and reduce the functions of NK cells and T lymphocytes [132]. Inselmann et al. noted that a higher CML-pDC count at diagnosis was associated with a poorer severity of the molecular response to nilotinib unless nilotinib therapy was combined with IFN (CML-V study), which strongly decreased the total number of circulating pDC, including CD86+ pDC [133]. Schütz [134] suggested that low levels of CD86+ on pDC may be a predictor of TFR as it increases RFS for patients with <95 CD86+ pDC compared to patients with> 95 CD86+ pDC per 105 lymphocytes (70 vs. 30.1%; p < 0.0001). Moreover, high levels of CD86+ pDC correlated with depletion of the leukemia-specific CD8+ T cell populations. In tumor immunotherapy using DC, cross-presentation of a sufficient amount of tumor antigens to effectively stimulate a CTL response may be of key importance [135]. This approach was used by Yang [136], who, in order to induce the cross-presentation of exogenous antigens derived from the BCR-ABL1 oncoprotein by DCs, was fused with cytoplasmic transduction peptides. In this study, the induced CTL showed a stronger ability to kill CML cells.
To date, the only curative therapy procedure for TKI non-responder CML patients remains alloHSCT. However, there are multiple pieces of evidence for the efficiency of immunotherapy treatment in CML, burdened with less mortality and morbidity. Previous studies compared the results of allogeneic bone marrow transfer with and without T cell depletion before the procedure. They found that the disposal of T cells from the graft results in a higher relapse rate in CML patients, suggesting an extremely important role of the T cell population in the eradication of leukemia cells [137]. Moreover, a clinical trial demonstrated that 40% of patients who discontinue imatinib did not show relapse symptoms that might manifest a T cell-mediated immune response with long-term memory [68].
Programmed death 1 (PD-1) is a member of CD28 co-stimulatory molecules, expressed on activated T and B cells, NK, and myeloid cells. PD-1 controls peripheral tolerance through binding to corresponding ligands PD-L1 and PD-L2, and inhibits proliferation, cytokine production, and cytotoxic function of effector cells. Recent studies demonstrated that elevated PD-1 expression on specific CTL directed against CML cells is related to loss of CTL lytic activity after co-stimulation with PD-L1-positive CML cells [138]. There was also evidence that PD-1 could be upregulated in CD4+ cells in CML patients [139]. Mumprecht et al. [140] proved the expression of PD-1 on CML-specific CTLs and the expression of PD-1 ligand (PD-L1) on CML cells in a mouse model, while extending the study to CML patients, who demonstrated increased PD-1 expression on their CD8+ T cells. These results suggest that PD-1 might contribute to maintaining immunity compromise during the chronic phase of the disease. Moreover, increased expression of PD-1 in CD8+ cells might be associated with disease progression and represents the mechanism of tumor escape from immunosurveillance [140]. Interestingly, Riether et al. [141] showed that PD-1 blockade in combination with immunotherapy could eradicate leukemic stem cells and decrease tumor burden in CML mice. However, there are no available data about alterations in PD-1 expression after discontinued imatinib treatment in CML patients. Investigations of the protein changes during the remission phase or in relapse may clarify if the blockade of the PD-1 molecule in CML could carry benefits in relapse patients.
Another modulator of the immune system that might influence maintaining TFR in CML patients is an infection of cytomegalovirus (CMV). CMV is the largest member of the virus family Herpesviridae and is a ubiquitous virus that infects almost all humans at some time in their lives. Symptomatic CMV infection predominantly occurs in immunocompromised hosts, such as patients after alloSCT, whereas symptomatic infection of healthy persons is rare [142]. There are relatively many studies showing an association of CMV reactivation with a reduced number of acute myeloid leukemia (AML) relapses following allo-SCT. Behrendt et al. [143] observed a significantly higher leukemic relapse risk after alloSCT myelodysplastic syndrome patients with a negative pre-transplantation CMV donor and recipient serostatus compared with patients with a positive pre-transplantation CMV serology of donors or recipients. In another study, Emaagacli et al. [144] reported that CMV reactivation is associated with a decreased risk of disease relapse in patients with AML. Furthermore, Green et al. [145] found that CMV reactivation in the first 100 days after transplantation is associated with a modest decrease in the risk of early relapse independent of acute GvHD in patients with AML. There are also studies in the literature on the favorable ‘virus-versus-leukemia’ effect from reactivating CMV in CML transplant recipients. Ito et al. [146] showed that CMV viral reactivation as a time-dependent covariate was an independent factor associated with a reduction in relapse in CML patients after allo-SCT. CMV reactivation may contribute to the beneficial effects of graft-versus-leukemia (GVL) by stimulating the immune system. It has been shown that subclinical CMV reactivation can lead to the expansion of specific subsets of NK and T cells with both antitumor and antiviral activity due to the common pathways of the immune response [147]. Kadowaki et al. [148] observed a relationship between CMV reactivation and expansion of LGL, which include CD8 + T lymphocytes, γδT lymphocytes, and NK cells. Additional analyses showed that NK cells in CMV + seropositive patients underwent phenotypic progression that was further enhanced by TKI, including dasatinib, in this study. Thus, CMV-associated NK cell activation may help to prevent CML recurrence upon discontinuation of treatment. The protective mechanism of CMV reactivation during relapse is unknown. There is a hypothesis that CMV reactivation might induce T cell and NK cell attack on malignant cells due to latent CMV virus presence or the activation of CTL affecting leukemic cells. NK cells play a crucial role in curtailing CMV and other viral infections in immunocompetent individuals through the expression of a series of activating receptors (NKG2D, DNAM-1, NKp46) that allow them to recognize and eliminate CMV and other virus-infected cells [142]. In in vitro experiments using IL-2-activated human NK cells, Iversen et al. [149] demonstrated that NK cells can inhibit CMV replication in CMV-infected fibroblasts by inducing IFN-beta release from infected fibroblasts. Kheav et al. [150] reported that a higher percentage of NKG2C+ NK cells is associated with a lower risk of acute CMV infection in patients undergoing solid organ transplantation or HSCT. Foley et al. [151] demonstrated increased populations of IFN-γ-producing NKG2C + NKG2A − NK cells in HSCT recipients after CMV viremia. TKI treatment induces a strong immune response that persists after treatment discontinuation, and which may also be antiviral. The research of Vigón et al. [152] demonstrated that seropositive for CMV, but without CMV viremia undergoing the TFR trial previously, stimulated with CMV peptides had an 8-fold increased expression of TCRγδ and produced over 18-fold more IFNγ from CD3+CD8+ T cells similar to CMV reactivated kidney transplant patients. However, the CML patients undergoing TFR did not reactivate CMV. Moreover, it was proved that γδT cells were able to recognize both CMV-infected cells and primary leukemic blasts; therefore, it has been proposed that γδ T cells recognizing CMV peptides cross-react with leukemia cells [153]. This may indicate that the presence of CMV along with leukemia cells and treatment with TKI may elicit a strong immune response against leukemia cells as well as virally infected cells. Therefore, it may be suggested that CMV together with TKIs modulate the immune system, so that immune control over the clone of malignant CML cells may be restored. The importance of CMV status in the donor and recipient is important to the success of the transplant. There is increasing evidence suggesting that stem cell transplant recipients receiving CMV seropositive donors show improved survival, event-free survival, reduced transplant-related mortality, or reduced risk of relapse [143,154]. However, studies emphasize that in CML patients, T cell depletion offsets the beneficial effects of donor status, suggesting that ultimately it is mediated by donor immunity. Nevertheless, it is difficult to clearly define to what extent CMV reactivation is a substitute for immunological factors that directly contribute to relapse.
Moreover, the immunomodulatory effect of TKIs promotes a strong cytotoxic response, which may be a breakthrough in the fight against HIV-1 infection. Salgado et al. [155] suggested that the primary antiviral mechanism against HIV-1 was based on inhibition of SAMHD1 phosphorylation in CD4+ T cells, which prevented ex vivo HIV-1 infection in CML patients treated with dasatinib. However, extending their research to the group of CML patients in TFR, they noticed that despite the unblocking of the SAMHD1 phosphorylation pathway due to the withdrawal of TKI, the frequency of proviral integration was more than 12-fold reduced [152]. This creates an opportunity for the transient use of TKIs in HIV-infected patients to modulate the immune response.
Studies on the immune status of CML patients in TFR were conducted by Hughes et al. [156], who showed that CML patients at diagnosis had impaired NK cell and CTL effector function that was restored by treatment when patients achieved MMR and DMR. More importantly, sufficient effector responses were maintained upon discontinuation of treatment in imatinib-treated patients with persistent DMR. It turns out that in the restoration of immune control over the leukemic clone, there is a depth of the molecular response achieved, because the cytolytic function of NK cells, and the decrease in Mo-MDSC (monocytic myeloid-derived suppressor cells) and PD-1 expression on CD4+ and CD8+ T cells was completely restored only in MR4.5 [156]. Maintaining a sustained anti-leukemic response and a successful TFR is accompanied by a decrease in the level of Treg [80] and an increase in the level of NK cells [127]. Immunological factors relevant to the success of TFR are summarized in Table 5.
Table 5.
Immune system factors critical to the success of TFR.
8. TKI Withdrawal Syndrome
The possibility to recover the proper function of the immune system during treatment with TKI and the restoration of immune control over a clone of malignant CML cells is most probably critical to achieve a long-term TFR. The importance of immunocompetent cells involved in the course of TKI discontinuation is also underlined by the presence of symptoms occurring in some patients with so-called “withdrawal syndrome” (WS). The etiology of this phenomenon remains elusive, but the hypothesis on its development involves cytokine release as a result of tyrosine kinase unblocking. TKI WS affects approximately 25–30% of patients after discontinuation of treatment [157,158] (Table 6). It usually presents as diffuse musculoskeletal pain from mild intensity relieved with common painkillers or non-steroidal anti-inflammatory drugs to moderate intensity requiring the use of corticosteroids [159]. The arms, hips, and extremities are the areas that are affected by the withdrawal syndrome [62,159,160]. In research on the risk of TKI withdrawal syndrome, Berger et al. [159] obtained a significantly higher incidence of WS after discontinuation of 2G TKI than with imatinib by analyzing a cohort of 427 patients from EURO-SKI and STIM2 studies. The pain may be related to the biological effects of inhibition of the c-Kit tyrosine kinase receptor through changes in sensitivity to thermal pain [161]. TKI WS may be caused by inflammation after TKI withdrawal due to the immunomodulatory effect of TKIs. However, most patients did not have elevated proinflammatory markers, including CRP protein, or the presence of autoimmune markers, which requires further studies [160]. Interestingly, a study in South Korea showed that itching is a manifestation of TKI withdrawal syndrome [162], which was not found in the analysis of patients in EURO-SKI and STIM2, which may highlight the differences in WS symptoms in patients from different ethnic groups [159,163].
Table 6.
TKI withdrawal syndrome.
9. Summary
According to the latest ELN recommendation from March 2020, TFR should be considered as a new goal for the treatment of CML, which has become a challenge on the way to curing CML. This approach is primarily aimed at improving the quality of life of CML patients and avoiding long-term toxicity of TKIs. The effectiveness of TFR tests reaches almost half of CML patients undertaking the TFR test. The maturing individualized approach to the patient allows for careful monitoring of the patient. Discontinuation of treatment should be specifically justified in patients with CML with stable DMR and in patients with a high priority for TFR, e.g., women wishing to become pregnant and younger patients, depending on whether the duration of treatment with both TKI and DMR is long enough. The immune mechanism that might be responsible for sustained TFR has not yet been clearly defined.
Author Contributions
Writing—original draft preparation, P.K.; writing—review and editing, K.G., P.K.; supervision, K.G.; funding acquisition, K.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Center in Poland, grant number NCN 2018/31/B/NZ6/03361.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973. [Google Scholar] [CrossRef]
- Bartram, C.R.; De Klein, A.; Hagemeijer, A.; Van Agthoven, T.; Van Kessel, A.G.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M.; et al. Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome inchronicmyelocytic leukaemia. Nature 1983. [Google Scholar] [CrossRef] [PubMed]
- Heisterkamp, N.; Stephenson, J.R.; Groffen, J.; Hansen, P.F.; De Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [CrossRef]
- Sasaki, K.; Strom, S.S.; O’Brien, S.; Jabbour, E.; Ravandi, F.; Konopleva, M.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Jain, P.; et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: Analysis of patient data from six prospective clinical trials. Lancet Haematol. 2015. [Google Scholar] [CrossRef]
- Thielen, N.; Visser, O.; Ossenkoppele, G.; Janssen, J. Chronic myeloid leukemia in the Netherlands: A population-based study on incidence, treatment, and survival in 3585 patients from 1989 to 2012. Eur. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M.L. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Welch, H.G.; Kramer, B.S.; Black, W.C. Epidemiologic Signatures in Cancer. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef]
- Hehlmann, R.; Lauseker, M.; Saußele, S.; Pfirrmann, M.; Krause, S.; Kolb, H.J.; Neubauer, A.; Hossfeld, D.K.; Nerl, C.; Gratwohl, A.; et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia 2017. [Google Scholar] [CrossRef]
- Hjorth-Hansen, H.; Stentoft, J.; Richter, J.; Koskenvesa, P.; Höglund, M.; Dreimane, A.; Porkka, K.; Gedde-Dahl, T.; Gjertsen, B.T.; Gruber, F.X.; et al. Safety and efficacy of the combination of pegylated interferon-α2b and dasatinib in newly diagnosed chronic-phase chronic myeloid leukemia patients. Leukemia 2016. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevärn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Björeman, M.; Flogegård, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011. [Google Scholar] [CrossRef] [PubMed]
- Preudhomme, C.; Guilhot, J.; Nicolini, F.E.; Guerci-Bresler, A.; Rigal-Huguet, F.; Maloisel, F.; Coiteux, V.; Gardembas, M.; Berthou, C.; Vekhoff, A.; et al. Imatinib plus Peginterferon Alfa-2a in Chronic Myeloid Leukemia. N. Engl. J. Med. 2010. [Google Scholar] [CrossRef] [PubMed]
- Eskazan, A.E.; Sadri, S.; Keskin, D.; Ayer, M.; Kantarcioglu, B.; Demirel, N.; Aydin, D.; Aydinli, F.; Yokus, O.; Ozunal, I.E.; et al. Outcomes of Chronic Myeloid Leukemia Patients With Early Molecular Response at 3 and 6 Months: A Comparative Analysis of Generic Imatinib and Glivec. Clin. Lymphoma Myeloma Leuk. 2017. [Google Scholar] [CrossRef] [PubMed]
- Malkan, U.Y.; Aksu, S.; Aktimur, S.H.; Atay, H.; Bektas, O.; Buyukasik, Y.; Demiroglu, H.; Eliacik, E.; Esme, M.; Hacihanefioglu, A.; et al. Generic imatinib mesylate is as effective as original glivec in the clinical management of CML. UHOD Uluslararasi Hematol. Derg. 2015. [Google Scholar] [CrossRef]
- Islamagic, E.; Hasic, A.; Kurtovic, S.; Suljovic Hadzimesic, E.; Mehinovic, L.; Kozaric, M.; Kurtovic-Kozaric, A. The Efficacy of Generic Imatinib as First- and Second-line Therapy: 3-Year Follow-up of Patients with Chronic Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sacha, T.; Góra-Tybor, J.; Szarejko, M.; Bober, G.; Grzybowska-Izydorczyk, O.; Niesiobędzka-Krężel, J.; Dudziński, M.; Wasilewska, E.; Myśliwiec, K.; Gil, J.; et al. A multicenter prospective study on efficacy and safety of imatinib generics: A report from Polish Adult Leukemia Group imatinib generics registry. Am. J. Hematol. 2017, 92, E125–E128. [Google Scholar] [CrossRef]
- Baccarani, M.; Castagnetti, F.; Gugliotta, G.; Rosti, G.; Soverini, S.; Albeer, A.; Pfirrmann, M. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. An international overview. Leukemia 2019, 33, 1173–1183. [Google Scholar] [CrossRef]
- Hughes, T.; Deininger, M.; Hochhaus, A.; Branford, S.; Radich, J.; Kaeda, J.; Baccarani, M.; Cortes, J.; Cross, N.C.P.; Druker, B.J.; et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: Review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006, 108, 28–37. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005. [Google Scholar] [CrossRef]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-Naïve chronic myeloid leukemia patients trial. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; Le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Müller, M.C.; Dietz, C.T.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia 2015. [Google Scholar] [CrossRef]
- Steegmann, J.L.; Baccarani, M.; Breccia, M.; Casado, L.F.; García-Gutiérrez, V.; Hochhaus, A.; Kim, D.W.; Kim, T.D.; Khoury, H.J.; Le Coutre, P.; et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia 2016, 30, 1648–1671. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Saglio, G.; Larson, R.A.; Kantarjian, H.M.; Kim, D.-W.; Issaragrisil, S.; Le Coutre, P.; Etienne, G.; Boquimpani, C.; Clark, R.E.; et al. Long-Term Outcomes in Patients with Chronic Myeloid Leukemia in Chronic Phase Receiving Frontline Nilotinib Versus Imatinib: Enestnd 10-Year Analysis. Blood 2019. [Google Scholar] [CrossRef]
- Gugliotta, G.; Castagnetti, F.; Breccia, M.; Levato, L.; Intermesoli, T.; D’adda, M.; Salvucci, M.; Stagno, F.; Rege Cambrin, G.; Tiribelli, M.; et al. Ten-Year Follow-up of Patients with Chronic Myeloid Leukemia Treated with Nilotinib in First-Line: Final Results of the Gimema CML 0307 Trial. Blood 2019, 134, 4145. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Kantarjian, H.M.; Brümmendorf, T.H.; Dyagil, I.; Griskevicius, L.; Malhotra, H.; Powell, C.; Gogat, K.; Countouriotis, A.M.; et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: Results from the BELA trial. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Massaro, F.; Molica, M.; Breccia, M. Ponatinib: A Review of Efficacy and Safety. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef]
- Cortes, J.E.; Hughes, T.P.; Mauro, M.J.; Hochhaus, A.; Rea, D.; Goh, Y.T.; Janssen, J.; Steegmann, J.L.; Heinrich, M.C.; Talpaz, M.; et al. Asciminib, a First-in-Class STAMP Inhibitor, Provides Durable Molecular Response in Patients (pts) with Chronic Myeloid Leukemia (CML) Harboring the T315I Mutation: Primary Efficacy and Safety Results from a Phase 1 Trial. Blood 2020, 136, 47–50. [Google Scholar] [CrossRef]
- Jiang, Q.; Huang, X.; Chen, Z.; Niu, Q.; Shi, D.; Li, Z.; Hou, Y.; Hu, Y.; Li, W.; Liu, X.; et al. Novel BCR-ABL1 Tyrosine Kinase Inhibitor (TKI) HQP1351 (Olverembatinib) Is Efficacious and Well Tolerated in Patients with T315I-Mutated Chronic Myeloid Leukemia (CML): Results of Pivotal (Phase II) Trials. Blood 2020, 136, 50–51. [Google Scholar] [CrossRef]
- Cortes, J.E.; Saikia, T.; Kim, D.-W.; Alvarado, Y.; Nicolini, F.E.; Khattry, N.; Rathnam, K.; Apperley, J.; Deininger, M.W.; de Lavallade, H.; et al. Phase 1 Trial of Vodobatinib, a Novel Oral BCR-ABL1 Tyrosine Kinase Inhibitor (TKI): Activity in CML Chronic Phase Patients Failing TKI Therapies Including Ponatinib. Blood 2020, 136, 51–52. [Google Scholar] [CrossRef]
- Hehlmann, R. The New ELN Recommendations for Treating CML. J. Clin. Med. 2020, 9, 3671. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome–positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Lomaia, E.; Turkina, A.; Moiraghi, B.; Sutton, M.U.; Pavlovsky, C.; Rojas, C.; Chuah, C.; Arthur, C.; Apperley, J.; et al. CML-114: Interim Analysis from the OPTIC Trial—A Dose-Ranging Study of 3 Starting Doses of Ponatinib. Clin. Lymphoma Myeloma Leuk. 2020, 20, S234. [Google Scholar] [CrossRef]
- Oehler, V.G. First-generation vs second-generation tyrosine kinase inhibitors: Which is best at diagnosis of chronic phase chronic myeloid leukemia? Hematology 2020. [Google Scholar] [CrossRef]
- Gratwohl, A.; Pfirrmann, M.; Zander, A.; Kröger, N.; Beelen, D.; Novotny, J.; Nerl, C.; Scheid, C.; Spiekermann, K.; Mayer, J.; et al. Long-term outcome of patients with newly diagnosed chronic myeloid leukemia: A randomized comparison of stem cell transplantation with drug treatment. Leukemia 2016. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.M.; Ghorab, A.; Sasaki, K.; Jabbour, E.J.; Nogueras Gonzalez, G.; Kanagal-Shamanna, R.; Issa, G.C.; Garcia-Manero, G.; Devendra, K.C.; et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer 2017. [Google Scholar] [CrossRef]
- Lübking, A.; Dreimane, A.; Sandin, F.; Isaksson, C.; Märkevärn, B.; Brune, M.; Ljungman, P.; Lenhoff, S.; Stenke, L.; Höglund, M.; et al. Allogeneic stem cell transplantation for chronic myeloid leukemia in the TKI era: Population-based data from the Swedish CML registry. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef]
- Cortes, J.; Rea, D.; Lipton, J.H. Treatment-free remission with first- and second-generation tyrosine kinase inhibitors. Am. J. Hematol. 2019, 94, 346–357. [Google Scholar] [CrossRef]
- Branford, S.; Fletcher, L.; Cross, N.C.P.; Müller, M.C.; Hochhaus, A.; Kim, D.W.; Radich, J.P.; Saglio, G.; Pane, F.; Kamel-Reid, S.; et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; White, H.E.; Müller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Branford, S.; Kamel-Reid, S.; Bendit, I.; Etienne, G.; Guerci-Bresler, A.; Hughes, T.P.; Lipton, J.H.; Leber, B.; Spector, N.; Steegmann, J.L.; et al. Early molecular response predicts achievement of undetectable BCR-ABL in patients (PTS) with chronic myeloid leukemia in chronic phase (CML-CP) treated with nilotinib: 3-year follow-up of ENESTCMR. Haematologica 2014, 99, 532. [Google Scholar]
- Pfirrmann, M.; Baccarani, M.; Saussele, S.; Guilhot, J.; Cervantes, F.; Ossenkoppele, G.; Hoffmann, V.S.; Castagnetti, F.; Hasford, J.; Hehlmann, R.; et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia 2016. [Google Scholar] [CrossRef]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Stagno, F.; Specchia, G.; Levato, L.; Martino, B.; D’Adda, M.; Abruzzese, E.; Pregno, P.; et al. The Use of EUTOS Long-Term Survival Score Instead of Sokal Score Is Strongly Advised in Elderly Chronic Myeloid Leukemia Patients. Blood 2018. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Bavaro, L.; de Benedittis, C.; Martelli, M.; Iurlo, A.; Orofino, N.; Sica, S.; Sorà, F.; Lunghi, F.; Ciceri, F.; et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Martelli, M.; Bavaro, L.; De Benedittis, C.; Iurlo, A.; Galimberti, S.; Pregno, P.; Bonifacio, M.; Lunghi, F.; Castagnetti, F.; et al. Detection of Actionable BCR-ABL1 Kinase Domain (KD) Mutations in Chronic Myeloid Leukemia (CML) Patients with Failure and Warning Response to Tyrosine Kinase Inhibitors (TKIs): Potential Impact of Next-Generation Sequencing (NGS) and Droplet Digital PCR. Blood 2019. [Google Scholar] [CrossRef]
- Hehlmann, R. Chronic Myeloid Leukemia in 2020. HemaSphere 2020, 4, e468. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X. Treatment-free remission in CML: Who, how, and why? Hematology 2017, 2017, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Abruzzese, E.; Accurso, V.; Albano, F.; Annunziata, M.; Barulli, S.; Beltrami, G.; Bergamaschi, M.; Binotto, G.; Bocchia, M.; et al. Managing chronic myeloid leukemia for treatment-free remission: A proposal from the GIMEMA CML WP. Blood Adv. 2019, 3, 4280–4290. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; White, H.E.; Colomer, D.; Ehrencrona, H.; Foroni, L.; Gottardi, E.; Lange, T.; Lion, T.; Machova Polakova, K.; Dulucq, S.; et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia 2015, 29, 999–1003. [Google Scholar] [CrossRef]
- Hughes, T.P.; Ross, D.M. Moving treatment-free remission into mainstream clinical practice in CML. Blood 2016, 128, 17–23. [Google Scholar] [CrossRef]
- Carlier, P.; Markarian, M.; Bernard, N.; Lagarce, L.; Dautriche, A.; Béné, J.; Sam-Lai, N.F.; Eftekhari, P. Erratum to: Pregnancy outcome among partners of male patients receiving imatinib, dasatinib or nilotinib in chronic myeloid leukemia: Reports collected by the French network pharmacovigilance centers. Arch. Gynecol. Obstet. 2017, 295, 269–271. [Google Scholar] [CrossRef]
- Shima, H.; Tokuyama, M.; Tanizawa, A.; Tono, C.; Hamamoto, K.; Muramatsu, H.; Watanabe, A.; Hotta, N.; Ito, M.; Kurosawa, H.; et al. Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. J. Pediatr. 2011. [Google Scholar] [CrossRef]
- Giona, F.; Siniscalchi, B.; Moleti, M.L.; Rea, M.; Marzella, D.; Nanni, M.; Gottardi, E.; Iori, A.P.; Diverio, D.; Testi, A.M.; et al. Can Children and Adolescents with Chronic Myelogenous Leukemia Be Cured Without Stem Cell Transplant? A Single Center Experience. Blood 2013, 122, 4033. [Google Scholar] [CrossRef]
- Bansal, D.; Shava, U.; Varma, N.; Trehan, A.; Marwaha, R.K. Imatinib has adverse effect on growth in children with chronic myeloid leukemia. Pediatr. Blood Cancer 2012, 59, 481–484. [Google Scholar] [CrossRef]
- Hijiya, N.; Schultz, K.R.; Metzler, M.; Millot, F.; Suttorp, M. Pediatric chronic myeloid leukemia is a unique disease that requires a different approach. Blood 2016, 127, 392–399. [Google Scholar] [CrossRef]
- Saußele, S.; Richter, J.; Hochhaus, A.; Mahon, F.X. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016, 30, 1638–1647. [Google Scholar] [CrossRef]
- Hochhaus, A. Educational session: Managing chronic myeloid leukemia as a chronic disease. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Charbonnier, A.; Cony-Makhoul, P.; Agape, P.; Nicolini, F.E.; Varet, B.; Gardembas, M.; Etienne, G.; Reá, D.; Roy, L.; et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J. Clin. Oncol. 2014, 32, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Rousselot, P.; Loiseau, C.; Delord, M.; Cayuela, J.M.; Spentchian, M. Late molecular recurrences in patients with chronic myeloid leukemia experiencing treatment-free remission. Blood Adv. 2020, 4, 3034–3040. [Google Scholar] [CrossRef]
- Cohen, J.; Palumbo, A.; Wing, J.; Heinrich, M.C. Case series of chronic myeloid leukemia patients who maintained deep molecular response (DMR) with very low-dose ponatinib: Experience in discontinuing low-dose ponatinib and treatment-free remission (TFR) outcomes. Leuk. Lymphoma 2020, 61, 2511–2514. [Google Scholar] [CrossRef]
- Etienne, G.; Guilhot, J.; Rea, D.; Rigal-Huguet, F.; Nicolini, F.; Charbonnier, A.; Guerci-Bresler, A.; Legros, L.; Varet, B.; Gardembas, M.; et al. Long-term follow-up of the French Stop Imatinib (STIM1) study in patients with chronic myeloid leukemia. J. Clin. Oncol. 2017, 35, 298–305. [Google Scholar] [CrossRef]
- Mahon, F.X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Ross, D.M.; Pagani, I.S.; Shanmuganathan, N.; Kok, C.H.; Seymour, J.F.; Mills, A.K.; Filshie, R.J.; Arthur, C.K.; Dang, P.; Saunders, V.A.; et al. Long-term treatment-free remission of chronic myeloid leukemia with falling levels of residual leukemic cells. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Yeung, D.T.; Dang, P.; Goyne, J.M.; Slader, C.; Filshie, R.J.; et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: Results from the TWISTER study. Blood J. Am. Soc. Hematol. 2013. [Google Scholar] [CrossRef]
- Mori, S.; le Coutre, P.; Abruzzese, E.; Martino, B.; Pungolino, E.; Elena, C.; Bergamaschi, M.; Assouline, S.; Di Bona, E.; Gozzini, A.; et al. Imatinib Suspension and Validation (ISAV) Study: Final Results at 79 Months. Blood 2018, 132, 461. [Google Scholar] [CrossRef]
- Zang, D.Y.; Lee, W.S.; Mun, Y.-C.; Do, Y.R.; Oh, S.; Lee, S.-E.; Choi, S.Y.; Kim, D.-W. Long-Term Follow-up after Treatment Discontinuation in Patients with Chronic Myeloid Leukemia: The Korean Imatinib Discontinuation (KID) Study. Blood 2018, 132, 4252. [Google Scholar] [CrossRef]
- Kim, D.D.H.; Bence-Bruckler, I.; Forrest, D.L.; Savoie, M.L.; Couban, S.; Busque, L.; Delage, R.; Laneuville, P.; Liew, E.; Xenocostas, A.; et al. Treatment-Free Remission Accomplished by Dasatinib (TRAD): Preliminary Results of the Pan-Canadian Tyrosine Kinase Inhibitor Discontinuation Trial. Blood 2016, 128, 1922. [Google Scholar] [CrossRef]
- Takahashi, N.; Nishiwaki, K.; Nakaseko, C.; Aotsuka, N.; Sano, K.; Ohwada, C.; Kuroki, J.; Kimura, H.; Tokuhira, M.; Mitani, K.; et al. Treatment-free remission after two-year consolidation therapy with nilotinib in patients with chronic myeloid leukemia: STAT2 trial in Japan. Haematologica 2018, 103, 1835–1842. [Google Scholar] [CrossRef]
- Hochhaus, A.; Masszi, T.; Giles, F.J.; Radich, J.P.; Ross, D.M.; Gómez Casares, M.T.; Hellmann, A.; Stentoft, J.; Conneally, E.; García-Gutiérrez, V.; et al. Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: Results from the ENESTfreedom study. Leukemia 2017, 31, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.M.; Masszi, T.; Gómez Casares, M.T.; Hellmann, A.; Stentoft, J.; Conneally, E.; Garcia-Gutierrez, V.; Gattermann, N.; le Coutre, P.D.; Martino, B.; et al. Durable treatment-free remission in patients with chronic myeloid leukemia in chronic phase following frontline nilotinib: 96-week update of the ENESTfreedom study. J. Cancer Res. Clin. Oncol. 2018, 144, 945–954. [Google Scholar] [CrossRef]
- Mahon, F.-X. ENESTop 5-Year Update: Durability of Treatment-Free Remission Following Second-Line Nilotinib and Exploratory Analysis of Molecular Response Regain after Nilotinib Re-Initiation in Patients with Chronic Myeloid Leukemia. Blood 2020, 136, 29–30. [Google Scholar] [CrossRef]
- Mahon, F.X.; Boquimpani, C.; Kim, D.W.; Benyamini, N.; Clementino, N.C.D.; Shuvaev, V.; Ailawadhi, S.; Lipton, J.H.; Turkina, A.G.; De Paz, R.; et al. Treatment-free remission after second-line nilotinib treatment in patients with chronic myeloid leukemia in chronic phase results from a single-group, phase 2, open-label study. Ann. Intern. Med. 2018, 168, 461–470. [Google Scholar] [CrossRef]
- Nagafuji, K.; Matsumura, I.; Shimose, T.; Kawaguchi, T.; Kuroda, J.; Nakamae, H.; Miyamoto, T.; Kadowaki, N.; Ishikawa, J.; Imamura, Y.; et al. Cessation of nilotinib in patients with chronic myelogenous leukemia who have maintained deep molecular responses for 2 years: A multicenter phase 2 trial, stop nilotinib (NILSt). Int. J. Hematol. 2019, 110, 675–682. [Google Scholar] [CrossRef]
- Okada, M.; Imagawa, J.; Tanaka, H.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Final 3-year Results of the Dasatinib Discontinuation Trial in Patients with Chronic Myeloid Leukemia Who Received Dasatinib as a Second-line Treatment. Clin. Lymphoma Myeloma Leuk. 2018, 18, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, J.; Tanaka, H.; Okada, M.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): A multicentre phase 2 trial. Lancet Haematol. 2015, 2, e528–e535. [Google Scholar] [CrossRef]
- Kimura, S.; Imagawa, J.; Murai, K.; Hino, M.; Kitawaki, T.; Okada, M.; Tanaka, H.; Shindo, M.; Kumagai, T.; Ikezoe, T.; et al. Treatment-free remission after first-line dasatinib discontinuation in patients with chronic myeloid leukaemia (first-line DADI trial): A single-arm, multicentre, phase 2 trial. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Kumagai, T.; Nakaseko, C.; Nishiwaki, K.; Yoshida, C.; Ohashi, K.; Takezako, N.; Takano, H.; Kouzai, Y.; Murase, T.; Matsue, K.; et al. Dasatinib cessation after deep molecular response exceeding 2 years and natural killer cell transition during dasatinib consolidation. Cancer Sci. 2018, 109, 182–192. [Google Scholar] [CrossRef]
- Shah, N.P.; García-Gutiérrez, V.; Jiménez-Velasco, A.; Larson, S.; Saussele, S.; Rea, D.; Mahon, F.X.; Levy, M.Y.; Gómez-Casares, M.T.; Pane, F.; et al. Dasatinib discontinuation in patients with chronic-phase chronic myeloid leukemia and stable deep molecular response: The DASFREE study. Leuk. Lymphoma 2020, 61, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Nicolini, F.E.; Tulliez, M.; Guilhot, F.; Guilhot, J.; Guerci-Bresler, A.; Gardembas, M.; Coiteux, V.; Guillerm, G.; Legros, L.; et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: Interim analysis of the STOP 2G-TKI study. Blood 2017, 129, 846–854. [Google Scholar] [CrossRef]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.J.W.M.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef]
- Clark, R.E.; Polydoros, F.; Apperley, J.F.; Milojkovic, D.; Rothwell, K.; Pocock, C.; Byrne, J.; de Lavallade, H.; Osborne, W.; Robinson, L.; et al. De-escalation of tyrosine kinase inhibitor therapy before complete treatment discontinuation in patients with chronic myeloid leukaemia (DESTINY): A non-randomised, phase 2 trial. Lancet Haematol. 2019, 6, e375–e383. [Google Scholar] [CrossRef]
- Legros, L.; Nicolini, F.E.; Etienne, G.; Rousselot, P.; Rea, D.; Giraudier, S.; Guerci-Bresler, A.; Huguet, F.; Gardembas, M.; Escoffre, M.; et al. Second tyrosine kinase inhibitor discontinuation attempt in patients with chronic myeloid leukemia. Cancer 2017, 123, 4403–4410. [Google Scholar] [CrossRef]
- Kim, D.D.H.; Busque, L.; Forrest, D.L.; Savoie, L.; Bence-Bruckler, I.; Couban, S.; Delage, R.; Xenocostas, A.; Liew, E.; Laneuville, P.; et al. Second Attempt of TKI Discontinuation with Dasatinib for Treatment-Free Remission after Failing First Attempt with Imatinib: Treatment-Free Remission Accomplished By Dasatinib (TRAD) Trial. Blood 2018, 132, 787. [Google Scholar] [CrossRef]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018. [Google Scholar] [CrossRef]
- Clark, R.E. Tyrosine Kinase Inhibitor Therapy Discontinuation for Patients with Chronic Myeloid Leukaemia in Clinical Practice. Curr. Hematol. Malig. Rep. 2019, 14, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Li, L.; Giannopoulos, K.; Chen, J.; Brunner, C.; Barth, T.; Schmitt, A.; Wiesneth, M.; Döhner, K.; Döhner, H.; et al. Chronic myeloid leukemia cells express tumor-associated antigens eliciting specific CD8+ T-cell responses and are lacking costimulatory molecules. Exp. Hematol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Bornhäuser, M.; Thiede, C.; Platzbecker, U.; Kiani, A.; Oelschlaegel, U.; Babatz, J.; Lehmann, D.; Hölig, K.; Radke, J.; Tuve, S.; et al. Prophylactic transfer of BCR-ABL-, PR1-, and WT1-reactive donor T cells after T cell-depleted allogeneic hematopoietic cell transplantation in patients with chronic myeloid leukemia. Blood 2011. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Li, L.; Ringhoffer, M.; Barth, T.F.E.; Giannopoulos, K.; Guillaume, P.; Ritter, G.; Wiesneth, M.; Döhner, H.; Schmitt, M. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8+ T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood 2005. [Google Scholar] [CrossRef]
- Quintarelli, C.; Dotti, G.; De Angelis, B.; Hoyos, V.; Mims, M.; Luciano, L.; Heslop, H.E.; Rooney, C.M.; Pane, F.; Savoldo, B. Cytotoxic T lymphocytes directed to the preferentially expressed antigen of melanoma (PRAME) target chronic myeloid leukemia. Blood 2008. [Google Scholar] [CrossRef]
- Qudaihi, G.A.; Lehe, C.; Dickinson, A.; Eltayeb, K.; Rasheed, W.; Chaudhri, N.; Aljurf, M.; Dermime, S. Identification of a novel peptide derived from the M-phase phosphoprotein 11 (MPP11) leukemic antigen recognized by human CD8+ cytotoxic T lymphocytes. Hematol. Oncol. Stem Cell Ther. 2010. [Google Scholar] [CrossRef]
- Ochi, T.; Fujiwara, H.; Suemori, K.; Azuma, T.; Yakushijin, Y.; Hato, T.; Kuzushima, K.; Yasukawa, M. Aurora-A kinase: A novel target of cellular immunotherapy for leukemia. Blood 2009. [Google Scholar] [CrossRef]
- Ureshino, H.; Shindo, T.; Kimura, S. Role of cancer immunology in chronic myelogenous leukemia. Leuk. Res. 2020, 88, 106273. [Google Scholar] [CrossRef]
- Hughes, A.; Yong, A.S.M. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front. Immunol. 2017, 8, 469. [Google Scholar] [CrossRef]
- Giannopoulos, K.; Dmoszynska, A.; Rolinski, J.; Greiner, J.; Stilgenbauer, S.; Schmitt, M. Identification of RHAMM-Derived CD8+ Restricted, Heteroclitical, Cryptic Epitope R9Y as a Promising Target for Immunotherapy of Chronic Lymphocytic Leukemia. Blood 2009. [Google Scholar] [CrossRef]
- Kanojia, D.; Garg, M.; Saini, S.; Agarwal, S.; Kumar, R.; Suri, A. Sperm associated antigen 9 expression and humoral response in chronic myeloid leukemia. Leuk. Res. 2010, 34, 858–863. [Google Scholar] [CrossRef]
- Hofmann, S.; Greiner, J. Immunogenic antigens as therapeutic targets against myeloid leukaemic cells. Leuk. Res. 2010, 34. [Google Scholar] [CrossRef]
- Matsushita, M.; Ozawa, K.; Suzuki, T.; Nakamura, M.; Nakano, N.; Kanchi, S.; Ichikawa, D.; Matsuki, E.; Sakurai, M.; Karigane, D.; et al. CXorf48 is a potential therapeutic target for achieving treatment-free remission in CML patients. Blood Cancer J. 2017, 7, 601. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Szmit, E.; Myśliwska, J.; Dobyszuk, A.; Myśliwski, A. CD4 +CD25 + T regulatory cells inhibit cytotoxic activity of T CD8 + and NK lymphocytes in the direct cell-to-cell interaction. Clin. Immunol. 2004. [Google Scholar] [CrossRef]
- Bachy, E.; Bernaud, J.; Roy, P.; Rigal, D.; Nicolini, F.E. Quantitative and functional analyses of CD4+CD25+FoxP3+ regulatory T cells in chronic phase chronic myeloid leukaemia patients at diagnosis and on imatinib mesylate. Br. J. Haematol. 2011, 153, 139–143. [Google Scholar] [CrossRef]
- Zahran, A.M.; Badrawy, H.; Ibrahim, A. Prognostic value of regulatory T cells in newly diagnosed chronic myeloid leukemia patients. Int. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Hus, I.; Tabarkiewicz, J.; Lewandowska, M.; Wasiak, M.; Wdowiak, P.; Kusz, M.; Legieć, M.; Dmoszyńska, A.; Roliński, J. Evaluation of monocyte-derived dendritic cells, T regulatory and Th17 cells in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Folia Histochem. Cytobiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Wang, L.; Owen, S.; Knight, K.; Watmough, S.J.; Clark, R.E. Naturally occurring CD4+ CD25+ FOXP3+ T-regulatory cells are increased in chronic myeloid leukemia patients not in complete cytogenetic remission and can be immunosuppressive. Exp. Hematol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Nadal, E.; Garin, M.; Kaeda, J.; Apperley, J.; Lechler, R.; Dazzi, F. Increased frequencies of CD4+CD25high Tregs correlate with disease relapse after allogeneic stem cell transplantation for chronic myeloid leukemia. Leukemia 2007. [Google Scholar] [CrossRef]
- Larmonier, N.; Janikashvili, N.; LaCasse, C.J.; Larmonier, C.B.; Cantrell, J.; Situ, E.; Lundeen, T.; Bonnotte, B.; Katsanis, E. Imatinib Mesylate Inhibits CD4 + CD25 + Regulatory T Cell Activity and Enhances Active Immunotherapy against BCR-ABL—Tumors. J. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, N.; Zhou, X.; Gao, G.; Li, L.; Huang, J.; Li, Y.; Lu, Q.; He, B.; Pan, C.; et al. Therapeutic immune monitoring of CD4+CD25+T cells in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Oncol. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.Y.; Yang, X.; Wei, Q.; Li, H.; Huang, X.B.; Wang, X.D. Effects of Tyrosine Kinase Inhibitors on the Th1 and Treg Cells of CML Patients. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2019, 27, 25–32. [Google Scholar] [CrossRef]
- Alves, R.; McArdle, S.E.B.; Vadakekolathu, J.; Gonçalves, A.C.; Freitas-Tavares, P.; Pereira, A.; Almeida, A.M.; Sarmento-Ribeiro, A.B.; Rutella, S. Flow cytometry and targeted immune transcriptomics identify distinct profiles in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitors with or without interferon-α. J. Transl. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sopper, S.; Mustjoki, S.; White, D.; Hughes, T.; Valent, P.; Burchert, A.; Gjertsen, B.T.; Gastl, G.; Baldauf, M.; Trajanoski, Z.; et al. Reduced CD62L expression on T cells and increased soluble CD62L levels predict molecular response to tyrosine kinase inhibitor therapy in early chronic-phase chronic myelogenous leukemia. J. Clin. Oncol. 2017, 35, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Mustjoki, S.; Auvinen, K.; Kreutzman, A.; Rousselot, P.; Hernesniemi, S.; Melo, T.; Lahesmaa-Korpinen, A.M.; Hautaniemi, S.; Bouchet, S.; Molimard, M.; et al. Rapid mobilization of cytotoxic lymphocytes induced by dasatinib therapy. Leukemia 2013. [Google Scholar] [CrossRef]
- Schiffer, C.A.; Cortes, J.E.; Hochhaus, A.; Saglio, G.; Le Coutre, P.; Porkka, K.; Mustjoki, S.; Mohamed, H.; Shah, N.P. Lymphocytosis after treatment with dasatinib in chronic myeloid leukemia: Effects on response and toxicity. Cancer 2016, 122, 1398–1407. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Kwaśnik, P.; Lemieszek, M.; Rzeski, W. Possibilities of using NK cells in cancer immunotherapy. Med. Ogólna Nauk. Zdrowiu 2020, 26, 8–16. [Google Scholar] [CrossRef]
- Chen, C.I.U.; Koschmieder, S.; Kerstiens, L.; Schemionek, M.; Altvater, B.; Pscherer, S.; Gerss, J.; Maecker, H.T.; Berdel, W.E.; Juergens, H.; et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia 2012. [Google Scholar] [CrossRef]
- Cayssials, E.; Guilhot, F. Chronic Myeloid Leukemia: Immunobiology and Novel Immunotherapeutic Approaches. BioDrugs 2017, 31, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kijima, M.; Gardiol, N.; Held, W. Natural killer cell mediated missing-self recognition can protect mice from primary chronic myeloid leukemia in vivo. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Boissel, N.; Rea, D.; Tieng, V.; Dulphy, N.; Brun, M.; Cayuela, J.-M.; Rousselot, P.; Tamouza, R.; Le Bouteiller, P.; Mahon, F.-X.; et al. BCR/ABL Oncogene Directly Controls MHC Class I Chain-Related Molecule A Expression in Chronic Myelogenous Leukemia. J. Immunol. 2006. [Google Scholar] [CrossRef]
- Ohyashiki, K.; Katagiri, S.I.; Tauchi, T.; Ohyashiki, J.H.; Maeda, Y.; Matsumura, I.; Kyo, T.I. Increased natural killer cells and decreased CD3 +CD8 +CD62L + T cells in CML patients who sustained complete molecular remission after discontinuation of imatinib. Br. J. Haematol. 2012, 102, 1368. [Google Scholar] [CrossRef]
- Mizoguchi, I.; Yoshimoto, T.; Katagiri, S.; Mizuguchi, J.; Tauchi, T.; Kimura, Y.; Inokuchi, K.; Ohyashiki, J.H.; Ohyashiki, K. Sustained upregulation of effector natural killer cells in chronic myeloid leukemia after discontinuation of imatinib. Cancer Sci. 2013. [Google Scholar] [CrossRef]
- Ménard, C.; Blay, J.Y.; Borg, C.; Michiels, S.; Ghiringhelli, F.; Robert, C.; Nonn, C.; Chaput, N.; Taïeb, J.; Delahaye, N.F.; et al. Natural killer cell IFN-γ levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematologica 2017, 102. [Google Scholar] [CrossRef] [PubMed]
- Ilander, M.; Olsson-Strömberg, U.; Schlums, H.; Guilhot, J.; Brück, O.; Lähteenmäki, H.; Kasanen, T.; Koskenvesa, P.; Söderlund, S.; Höglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Rossignol, A.; Levescot, A.; Jacomet, F.; Robin, A.; Basbous, S.; Giraud, C.; Roy, L.; Guilhot, F.; Turhan, A.G.; Barra, A.; et al. Evidence for BCR-ABL-dependent dysfunctions of iNKT cells from chronic myeloid leukemia patients. Eur. J. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Seggewiss, R.; Price, D.A.; Purbhoo, M.A. Immunomodulatory effects of imatinib and second-generation tyrosine kinase inhibitors on T cells and dendritic cells: An update. Cytotherapy 2008. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Luppi, M.; Barozzi, P.; Quadrelli, C.; Basso, S.; Vallerini, D.; Zanetti, E.; Morselli, M.; Forghieri, F.; Maccaferri, M.; et al. Emergence of BCR-ABL-specific cytotoxic T cells in the bone marrow of patients with Ph+ acute lymphoblastic leukemia during long-term imatinib mesylate treatment. Blood 2010. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Schmitt, A.; Giannopoulos, K.; Chen, B.; Rojewski, M.; Döhner, H.; Bunjes, D.; Schmitt, M. Imatinib impairs the proliferation and function of CD4+CD25 + regulatory T cells in a dose-dependent manner. Int. J. Oncol. 2007, 31, 1133–1139. [Google Scholar]
- Ochando, J.C.; Homma, C.; Yang, Y.; Hidalgo, A.; Garin, A.; Tacke, F.; Angeli, V.; Li, Y.; Boros, P.; Ding, Y.; et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 2006. [Google Scholar] [CrossRef]
- Inselmann, S.; Wang, Y.; Saussele, S.; Fritz, L.; Schutz, C.; Huber, M.; Liebler, S.; Ernst, T.; Cai, D.; Botschek, S.; et al. Development, function, and clinical significance of plasmacytoid dendritic cells in chronic myeloid leukemia. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schütz, C.; Inselmann, S.; Sausslele, S.; Dietz, C.T.; Müller, M.C.; Eigendorff, E.; Brendel, C.A.; Metzelder, S.K.; Brümmendorf, T.H.; Waller, C.; et al. Expression of the CTLA-4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; Van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, H.; Huang, Z.; Tao, K.; Huang, N.; Peng, Z.; Feng, W. Induction of CML-specific immune response through cross-presentation triggered by CTP-mediated BCR-ABL-derived peptides. Cancer Lett. 2020. [Google Scholar] [CrossRef] [PubMed]
- Drobyski, W.R.; Hessner, M.J.; Klein, J.P.; Kabler-Babbitt, C.; Vesole, D.H.; Keever-Taylor, C.A. T-cell depletion plus salvage immunotherapy with donor leukocyte infusions as a strategy to treat chronic-phase chronic myelogenous leukemia patients undergoing HLA-identical sibling marrow transplantation. Blood 1999. [Google Scholar] [CrossRef]
- Matte-Martone, C.; Venkatesan, S.; Tan, H.S.; Athanasiadis, I.; Chang, J.; Pavisic, J.; Shlomchik, W.D. Graft-versus-Leukemia (GVL) against Mouse Blast-Crisis Chronic Myelogenous Leukemia (BC-CML) and Chronic-Phase Chronic Myelogenous Leukemia (CP-CML): Shared Mechanisms of T Cell Killing, but Programmed Death Ligands Render CP-CML and Not BC-CML GVL Resist. J. Immunol. 2011. [Google Scholar] [CrossRef]
- Christiansson, L.; Söderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S.I. Increased Level of Myeloid-Derived Suppressor Cells, Programmed Death Receptor Ligand 1/Programmed Death Receptor 1, and Soluble CD25 in Sokal High Risk Chronic Myeloid Leukemia. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Mumprecht, S.; Schürch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood 2009. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Gschwend, T.; Huguenin, A.L.; Schürch, C.M.; Ochsenbein, A.F. Blocking programmed cell death 1 in combination with adoptive cytotoxic T-cell transfer eradicates chronic myelogenous leukemia stem cells. Leukemia 2015, 29, 1781–1785. [Google Scholar] [CrossRef]
- Magri, G.; Muntasell, A.; Romo, N.; Sáez-Borderías, A.; Pende, D.; Geraghty, D.E.; Hengel, H.; Angulo, A.; Moretta, A.; López-Botet, M. NKp46 and DNAM-1 NK-cell receptors drive the response to human cytomegalovirus-infected myeloid dendritic cells overcoming viral immune evasion strategies. Blood 2011. [Google Scholar] [CrossRef]
- Behrendt, C.E.; Rosenthal, J.; Bolotin, E.; Nakamura, R.; Zaia, J.; Forman, S.J. Donor and Recipient CMV Serostatus and Outcome of Pediatric Allogeneic HSCT for Acute Leukemia in the Era of CMV-Preemptive Therapy. Biol. Blood Marrow Transplant. 2009. [Google Scholar] [CrossRef]
- Elmaagacli, A.H.; Steckel, N.K.; Koldehoff, M.; Hegerfeldt, Y.; Trenschel, R.; Ditschkowski, M.; Christoph, S.; Gromke, T.; Kordelas, L.; Ottinger, H.D.; et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: Evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood 2011. [Google Scholar] [CrossRef] [PubMed]
- Green, M.L.; Leisenring, W.M.; Xie, H.; Walter, R.B.; Mielcarek, M.; Sandmaier, B.M.; Riddell, S.R.; Boeckh, M. CMV reactivation after allogeneic HCT and relapse risk: Evidence for early protection in acute myeloid leukemia. Blood 2013. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Pophali, P.; Co, W.; Koklanaris, E.K.; Superata, J.; Fahle, G.A.; Childs, R.; Battiwalla, M.; Barrett, A.J. CMV reactivation is associated with a lower incidence of relapse after allo-SCT for CML. Bone Marrow Transplant. 2013, 48, 1313–1316. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Climent, N.; Plana, M. Immunomodulatory activity of tyrosine kinase inhibitors to elicit cytotoxicity against cancer and viral infection. Front. Pharmacol. 2019, 10, 1232. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ishiyama, K.; Kitawaki, T. Cytomegalovirus pulls strings behind NK cells. Oncotarget 2017, 8, 93297–93298. [Google Scholar] [CrossRef]
- Iversen, A.-C.; Norris, P.S.; Ware, C.F.; Benedict, C.A. Human NK Cells Inhibit Cytomegalovirus Replication through a Noncytolytic Mechanism Involving Lymphotoxin-Dependent Induction of IFN-β. J. Immunol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Kheav, V.D.; Busson, M.; Scieux, C.; de Latour, R.P.; Maki, G.; Haas, P.; Mazeron, M.C.; Carmagnat, M.; Masso, E.; Xhaard, A.; et al. Favorable impact of natural killer cell reconstitution on chronic graft-versus-host disease and cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation. Haematologica 2014. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Vergès, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C + natural killer cells with potent function. Blood 2012. [Google Scholar] [CrossRef] [PubMed]
- Vigón, L.; Rodríguez-Mora, S.; Luna, A.; Sandonís, V.; Mateos, E.; Bautista, G.; Steegmann, J.L.; Climent, N.; Plana, M.; Pérez-Romero, P.; et al. Cytotoxic cell populations developed during treatment with tyrosine kinase inhibitors protect autologous CD4+ T cells from HIV-1 infection. Biochem. Pharmacol. 2020, 182. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Van Dorp, S.; Kersting, S.; Pietersma, F.; Lindemans, C.; Hol, S.; Heijhuurs, S.; Sebestyen, Z.; Gründer, C.; Marcu-Malina, V.; et al. γδT cells elicited by CMV reactivation after allo-SCT cross-recognize CMV and leukemia. Leukemia 2013, 27, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Brand, R.; Einsele, H.; Frassoni, F.; Niederwieser, D.; Cordonnier, C. Donor CMV serologic status and outcome of CMV-seropositive recipients after unrelated donor stem cell transplantation: An EBMT megafile analysis. 2003. [Google Scholar] [CrossRef]
- Salgado, M.; Martinez-Picado, J.; Gálvez, C.; Rodríguez-Mora, S.; Rivaya, B.; Urrea, V.; Mateos, E.; Alcamí, J.; Coiras, M. Dasatinib protects humanized mice from acute HIV-1 infection. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.L.; Hughes, T.P.; Yong, A.S.M. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef]
- Sweet, K. Starting Tyrosine Kinase Inhibitor Cessation in Chronic-Phase Chronic Myeloid Leukemia Patient. Hematology 2018, 15. [Google Scholar] [CrossRef]
- Berger, M.G.; Pereira, B.; Oris, C.; Saugues, S.; Cony-Makhoul, P.; Gardembas, M.; Legros, L.; Escoffre-Barbe, M.; Nicolini, F.E.; Rousselot, P.; et al. Osteoarticular Pain after Discontinuation of Tyrosine Kinase Inhibitors (TKI): A French Cohort. Blood 2015, 126, 137. [Google Scholar] [CrossRef]
- Berger, M.G.; Pereira, B.; Rousselot, P.; Cony-Makhoul, P.; Gardembas, M.; Legros, L.; Escoffre-Barbe, M.; Nicolini, F.E.; Saugues, S.; Lambert, C.; et al. Longer treatment duration and history of osteoarticular symptoms predispose to tyrosine kinase inhibitor withdrawal syndrome. Br. J. Haematol. 2019. [Google Scholar] [CrossRef]
- Richter, J.; Söderlund, S.; Lübking, A.; Dreimane, A.; Lotfi, K.; Markevärn, B.; Själander, A.; Saussele, S.; Olsson-Strömberg, U.; Stenke, L. Musculoskeletal pain in patients with chronic myeloid leukemia after discontinuation of imatinib: A tyrosine kinase inhibitor withdrawal syndrome? J. Clin. Oncol. 2014, 32, 2821–2823. [Google Scholar] [CrossRef]
- Ceko, M.; Milenkovic, N.; Le Coutre, P.; Westermann, J.; Lewin, G.R. Inhibition of c-Kit signaling is associated with reduced heat and cold pain sensitivity in humans. Pain 2014. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Choi, S.Y.; Song, H.Y.; Kim, S.H.; Choi, M.Y.; Park, J.S.; Kim, H.J.; Kim, S.H.; Zang, D.Y.; Oh, S.; et al. Imatinib withdrawal syndrome and longer duration of imatinib have a close association with a lower molecular relapse after treatment discontinuation: The KID study. Haematologica 2016, 101, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Tauchi, T.; Saito, Y.; Suguro, T.; Asano, M.; Yoshizawa, S.; Sakuta, J.; Akahane, D.; Tanaka, Y.; Furuya, N.; et al. Musculoskeletal pain after stopping tyrosine kinase inhibitor in patients with chronic myeloid leukemia: A questionnaire survey. Rinsho Ketsueki 2016. [Google Scholar] [CrossRef]
- Takahashi, N.; Tauchi, T.; Kitamura, K.; Miyamura, K.; Saburi, Y.; Hatta, Y.; Miyata, Y.; Kobayashi, S.; Usuki, K.; Matsumura, I.; et al. Deeper molecular response is a predictive factor for treatment-free remission after imatinib discontinuation in patients with chronic phase chronic myeloid leukemia: The JALSG-STIM213 study. Int. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Vagge, E.; le Coutre, P.; Abruzzese, E.; Martino, B.; Pungolino, E.; Elena, C.; Pierri, I.; Assouline, S.; D’Emilio, A.; et al. Age and dPCR can predict relapse in CML patients who discontinued imatinib: The ISAV study. Am. J. Hematol. 2015, 90, 910–914. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).