
The Relationship between Epigenetic Age and Myocardial Infarction/Acute Coronary Syndrome in a Population-Based Nested Case-Control Study

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Study Population and Design
2.2. Sample Selection Process
2.3. Ethics
2.4. Data Collection
2.5. DNAm Profiling
2.6. Data Preprocessing and Quality Control (QC)
2.7. DNAm Age Calculation
2.8. Statistical Analysis
3. Results
3.1. Cases and Controls Have Significant Differences in Basic Phenotype Characteristics
3.2. Association between Age Acceleration and Risk of MI/ACS
4. Discussion
5. Study Limitations and Advantages
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations, Department of Economic and Social Affairs, Population Division (2017). World Population Prospects: The 2017 Revision, Key Findings and Advance Tables. Working Paper No. ESA/P/WP/248. Available online: https://population.un.org/wpp/Publications/Files/WPP2017_KeyFindings.pdf (accessed on 29 October 2021).
- WHO/Global Status Report on Noncommunicable Diseases. 2014. Available online: https://www.who.int/publications/i/item/9789241564854 (accessed on 29 October 2021).
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D.; Martienssen, R.F.; Russo, V.E.A. Introduction. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996. [Google Scholar]
- Jones, M.J.; Goodman, S.J.; Kobor, M. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; Van Eijk, K.; Van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2012, 49, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A.T.; Levine, M.E.; Boks, M.P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 2021, 69, 101348. [Google Scholar] [CrossRef]
- Lee, Y.; Haftorn, K.L.; Denault, W.R.P.; Nustad, H.E.; Page, C.M.; Lyle, R.; Lee-Odegard, S.; Moen, G.-H.; Prasad, R.B.; Groop, L.C.; et al. Blood-based epigenetic estimators of chronological age in human adults using DNA methylation data from the Illumina MethylationEPIC array. BMC Genom. 2020, 21, 747. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Shireby, G.L.; Davies, J.P.; Francis, P.T.; Burrage, J.; Walker, E.M.; Neilson, G.W.A.; Dahir, A.; Thomas, A.J.; Love, S.; Smith, R.G.; et al. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 2020, 143, 3763–3775. [Google Scholar] [CrossRef]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.; Aviv, A.; McGue, M.; Christensen, K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 2015, 15, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Perna, L.; Zhang, Y.; Mons, U.; Holleczek, B.; Saum, K.-U.; Brenner, H. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenet. 2016, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugué, P.-A.; Bassett, J.K.; Joo, J.H.E.; Baglietto, L.; Jung, C.-H.; Wong, E.M.; Fiorito, G.; Schmidt, D.; Makalic, E.; Li, S.; et al. Association of DNA Methylation-Based Biological Age With Health Risk Factors and Overall and Cause-Specific Mortality. Am. J. Epidemiol. 2018, 187, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [Green Version]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin. Epigenet. 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Gurven, M.; Levine, M.E.; Trumble, B.C.; Kaplan, H.; Allayee, H.; Ritz, B.R.; Chen, B.; Lu, A.T.; Rickabaugh, T.M.; et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016, 17, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EUROSTAT. Mortality and Life Expectancy Statistics. 2021. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Mortality_and_life_expectancy_statistics (accessed on 30 October 2021).
- Global Age Watch Index 2015: Insight report. Available online: https://www.helpage.org/global-agewatch/reports/global-agewatch-index-2015-insight-report-summary-and-methodology/ (accessed on 29 October 2021).
- Bobak, M.; Malyutina, S.; Horvat, P.; Pajak, A.; Tamosiunas, A.; Kubinova, R.; Simonova, G.; Topor-Madry, R.; Peasey, A.; Pikhart, H.; et al. Alcohol, drinking pattern and all-cause, cardiovascular and alcohol-related mortality in Eastern Europe. Eur. J. Epidemiol. 2016, 31, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Tillmann, T.; Pikhart, H.; Peasey, A.; Kubínová, R.; Pajak, A.; Tamosiūnas, A.; Malyutina, S.; Steptoe, A.; Kivimäki, M.; Marmot, M.; et al. Psychosocial and socioeconomic determinants of cardiovascular mortality in Eastern Europe: A multicentre prospective cohort study. PLoS Med. 2017, 14, e1002459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefler, D.; Brett, D.; Sarkadi-Nagy, E.; Kopczynska, E.; Detchev, S.; Bati, A.; Scrob, M.; Koenker, D.; Aleksov, B.; Douarin, E.; et al. Traditional Eastern European diet and mortality: Prospective evidence from the HAPIEE study. Eur. J. Nutr. 2021, 60, 1091–1100. [Google Scholar] [CrossRef]
- Ryabikov, A.; Maksimov, V.; Holmes, M.; Bobak, M.; Malyutina, S. The association between epigenetic age and progression of subclinical atherosclerosis in ageing cohort. Atheroscler 2020, 315, e132. [Google Scholar] [CrossRef]
- Peasey, A.; Bobak, M.; Kubinova, R.; Malyutina, S.; Pajak, A.; Tamosiunas, A.; Pikhart, H.; Nicholson, A.; Marmot, M. Determinants of cardiovascular disease and other non-communicable diseases in Central and Eastern Europe: Rationale and design of the HAPIEE study. BMC Public Health 2006, 6, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.-P.; Triche, T.J., Jr.; Hansen, K.D. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2016, 33, 558–560. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Langie, S.A.S.; De Boever, P.; Taylor, J.A.; Niu, L.; De Boever, P. RELIC: A novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genom. 2017, 18, 4. [Google Scholar] [CrossRef] [Green Version]
- Chervova, O.; Conde, L.; Guerra-Assunção, J.A.; Moghul, I.; Webster, A.P.; Berner, A.; Cadieux, E.L.; Tian, Y.; Voloshin, V.; Jesus, T.F.; et al. The Personal Genome Project-UK, an open access resource of human multi-omics data. Sci. Data 2019, 6, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fix, E.; Hodges, J.L., Jr. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev. 1989, 57, 238–247. [Google Scholar] [CrossRef]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.; Wrigglesworth, J.; Loong, J.; Fransquet, P.; Woods, R.L. A Systematic Review and Meta-analysis of Environmental, Lifestyle, and Health Factors Associated With DNA Methylation Age. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2020, 75, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.; Shah, S.; McRae, A.; Ritchie, S.J.; Terrera, G.M.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Chen, B.H.; Hernandez, D.G.; Singleton, A.B.; Ferrucci, L.; Bandinelli, S.; Salfati, E.; Manson, J.E.; Quach, A.; et al. Menopause accelerates biological aging. Proc. Natl. Acad. Sci. USA 2016, 113, 9327–9332. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ni, W.; Yao, Y.; Just, A.; Heiss, J.; Wei, Y.; Gao, X.; Coull, B.A.; Kosheleva, A.; Baccarelli, A.A.; et al. DNA methylation-based biomarkers of age acceleration and all-cause death, myocardial infarction, stroke, and cancer in two cohorts: The NAS, and KORA F4. EBioMedicine 2021, 63, 103151. [Google Scholar] [CrossRef] [PubMed]
- Roetker, N.S.; Pankow, J.; Bressler, J.; Morrison, A.C.; Boerwinkle, E. Prospective Study of Epigenetic Age Acceleration and Incidence of Cardiovascular Disease Outcomes in the ARIC Study (Atherosclerosis Risk in Communities). Circ. Genom. Precis. Med. 2018, 11, e001937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.; Tsai, P.-C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [Green Version]
- Pagiatakis, C.; Musolino, E.; Gornati, R.; Bernardini, G.; Papait, R. Epigenetics of aging and disease: A brief overview. Aging Clin. Exp. Res. 2021, 33, 737–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Sanlés, A.; Baixeras, S.S.; Subirana, I.; Degano, I.R.; Elosua, R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: A systematic review. Atherosclerosis 2017, 263, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Curcio, S.; Subirana, I.; Marrugat, J.; Elosua, R. DNA Methylation and Age-Independent Cardiovascular Risk, an Epigenome-Wide Approach. Arter. Thromb. Vasc. Biol. 2018, 38, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Jaramillo, V.; Portilla-Fernandez, E.; Glisic, M.; Voortman, T.; Bramer, W.; Chowdhury, R.; Roks, A.J.; Danser, A.H.J.; Muka, T.; Nano, J.; et al. The role of DNA methylation and histone modifications in blood pressure: A systematic review. J. Hum. Hypertens. 2019, 33, 703–715. [Google Scholar] [CrossRef]
- Hartman, R.J.G.; Huisman, S.; Ruijter, H.M.D. Sex differences in cardiovascular epigenetics-a systematic review. Biol. Sex Differ. 2018, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Brewer, A.; Bell, J.T. DNA methylation signatures of incident coronary heart disease: Findings from epigenome-wide association studies. Clin. Epigenet. 2021, 13, 186. [Google Scholar] [CrossRef]
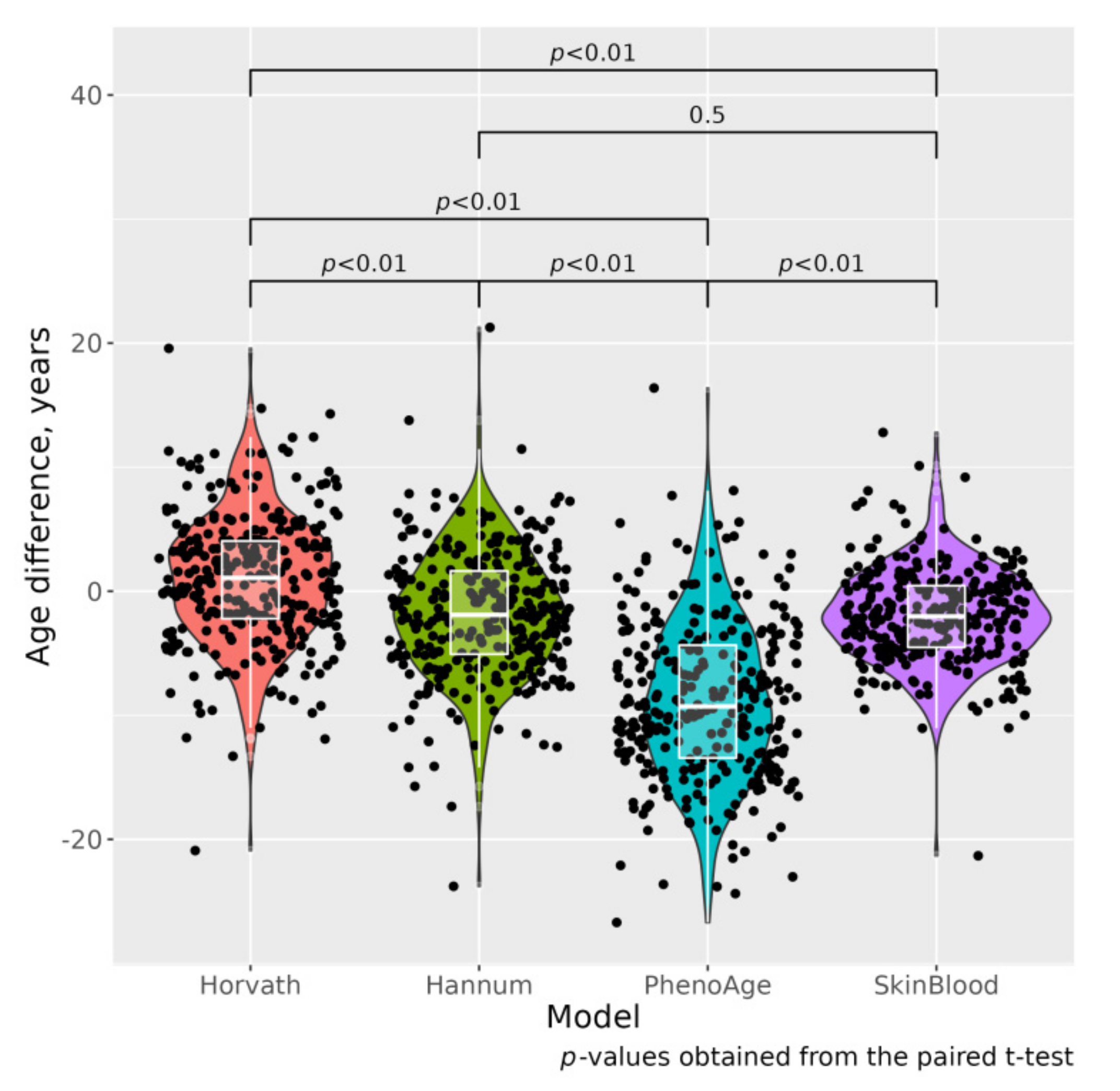
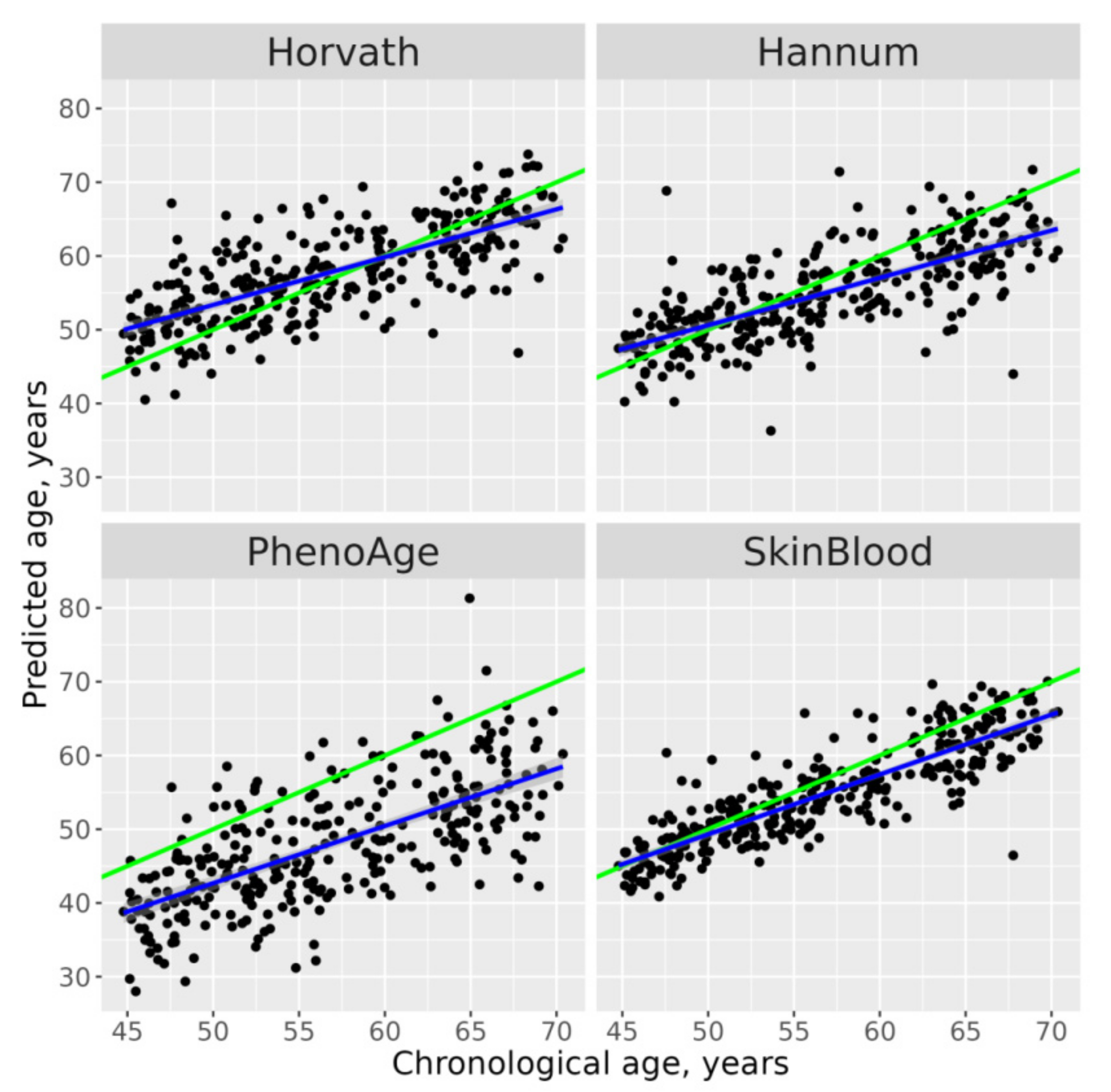

{kind=link}
{kind=link}
| Covariates | Cases (Incident MI/ACS) | Controls | p-Value a |
|---|---|---|---|
| Observed | 129 | 177 | |
| Age at baseline, years (mean, SD) | 59.8 (6.87) | 54.5 (6.45) | <0.001 |
| Females (%) | 62 (48.1) | 73 (58.8) | 0.064 |
| Systolic blood pressure, mmHg (mean, SD) | 151.6 (26.93) | 133.2 (21.87) | <0.001 |
| Diastolic blood pressure, mmHg (mean, SD) | 92.3 (14.36) | 86.0 (12.69) | <0.001 |
| Body mass index, kg/sqm (mean, SD) | 28.8 (5.73) | 27.50 (4.90) | 0.031 |
| Waist/hip ratio, unit (mean, SD) | 0.90 (0.077) | 0.87 (0.087) | 0.002 |
| Total cholesterol mmol/L (mean, SD) | 6.61 (1.27) | 6.42 (1.28) | 0.204 |
| LDL cholesterol, mmol/L (mean, SD) | 4.32 (1.14) | 4.15 (1.13) | 0.207 |
| Glucose, plasma, mmol/L mean, SD) | 6.41 (2.29) | 5.77 (0.85) | 0.001 |
| Hypertension (%) | 96 (74.4) | 80 (45.2) | <0.001 |
| HT treatment (among HT), (%) | 46 (47.9) | 46 (27.5) | 0.006 |
| Diabetes mellitus type 2 (%) | 24 (18.9) | 10 (5.8) | <0.001 |
| DM2 treatment (among DM2), (%) | 8 (33.3) | 3.(30.0) | 0.850 |
| Frequency of drinking (%) Non-drinkers | 24 (18.6) | 15 (8.5) | 0.050 |
| <1/month | 55 (42.6) | 76 (42.9) | |
| 1–3/month | 25 (19.4) | 35 (19.8) | |
| 1–4/week | 22 (17.1) | 48 (27.1) | |
| 5+/week | 3 (2.3) | 3 (1.7) | |
| Smoking (%) Never smoked | 75 (58.1) | 105 (59.3) | 0.066 |
| Former smoking | 10 (7.8) | 27 (15.3) | |
| Present smoker | 44 (34.1) | 56 (31.6) | |
| Married (%) | 96 (74.4) | 135 (76.3) | 0.405 |
| University education (%) | 27 (20.9) | 56 (31.6) | <0.001 |
| Difference EA–chronological age by four measures: | |||
| ΔAHr, year | 0.055 (5.35) | 1.663 (5.09) | 0.008 |
| ΔAHn, year | −2.702 (5.36) | −1.161 (4.82) | 0.009 |
| ΔAPh, year | −8.945 (6.43) | −8.762 (6.38) | 0.806 |
| ΔASB, year | −2.551 (4.06) | −1.550 (3.58) | 0.023 |
| Measure of Epigenetic Age | n, Case/Control | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|
| OR (95% CI) | OR (95% CI) | OR (95% CI) | OR (95% CI) | ||
| ΔAHr, per 1 year | 129/177 | 1.016 (0.96–1.07) | 1.003 (0.87–1.36) | 1.008 (0.95–1.06) | 1.009 (0.95–1.07) |
| p-value for trends | 0.563 | 0.911 | 0.785 | 0.763 | |
| ΔAHn, per 1 year | 129/177 | 1.023 (0.95–1.08) | 1.001 (0.95–1.06) | 1.006 (0.95–1.07) | 1.012 (0.95–1.08) |
| p-value for trends | 0.418 | 0.961 | 0.842 | 0.708 | |
| ΔAPh, per 1 year | 129/177 | 1.032 (0.99–1.07) | 1.021 (0.98–1.06) | 1.017 (0.98–1.06) | 1.017 (0.97–1.06) |
| p-value for trends | 0.126 | 0.310 | 0.430 | 0.459 | |
| ΔASB, per 1 year | 129/177 | 1.002 (0.94–1.07) | 0.991 (0.93–1.06) | 0.997 (0.93–1.07) | 1.009 (0.93–1.09) |
| p-value for trends | 0.962 | 0.802 | 0.927 | 0.825 | |
| Measure of Epigenetic Age | n, Case/Control | Tertiles | Absolute Difference T1-T2 T2-T3 | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|---|---|
| OR (95% CI) | OR (95% CI) | OR (95% CI) | OR (95% CI) | ||||
| ΔAHr, year | 129/177 | T1 (ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 5.64 | 0.89 (0.49–1.63) | 0.83 (0.45–1.53) | 0.83 (0.44–1.54) | 0.91 (0.47–1.77) | ||
| T3 | 5.48 | 1.26 (0.65–2.44) | 1.14 (0.59–2.22) | 1.21 (0.61–2.40) | 1.24 (0.60–2.56) | ||
| p-value for trends | 0.510 | 0.738 | 0.624 | 0.593 | |||
| ΔAHn, year | 129/177 | T1 (ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 5.35 | 1.28 (0.68–2.39) | 1.20 (0.63–2.24) | 1.26 (0.66–2.40) | 1.22 (0.61–2.44) | ||
| T3 | 5.40 | 1.57 (0.79–3.14) | 1.26 (0.61–2.60) | 1.36 (0.65–2.85) | 1.36 (0.63–2.96) | ||
| p-value for trends | 0.198 | 0.526 | 0.408 | 0.437 | |||
| ΔAPh, year | 129/177 | T1 (ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 6.49 | 1.19 (0.64–2.21) | 1.18 (0.63–2.20) | 1.21 (0.65–2.28) | 1.17 (0.61–2.27) | ||
| T3 | 7.40 | 2.09 (1.11–3.94) | 1.84 (0.99–3.52) | 1.78 (0.92–3.43) | 1.64 (0.82–3.31) | ||
| p-value for trends | 0.022 | 0.065 | 0.088 | 0.171 | |||
| ΔASB, year | 129/177 | T1 (ref) | 1.0 | 1.0 | 1.0 | 1.0 | |
| T2 | 3.94 | 0.88 (0.47–1.62) | 0.80 (0.43–1.51) | 0.84 (0.45–1.58) | 0.99 (0.50–1.94) | ||
| T3 | 4.06 | 1.13 (0.60–2.11) | 1.00 (0.53–1.89) | 1.09 (0.57–2.09) | 1.18 (0.60–2.37) | ||
| p-value for trends | 0.699 | 0.948 | 0.738 | 0.637 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyutina, S.; Chervova, O.; Tillmann, T.; Maximov, V.; Ryabikov, A.; Gafarov, V.; Hubacek, J.A.; Pikhart, H.; Beck, S.; Bobak, M. The Relationship between Epigenetic Age and Myocardial Infarction/Acute Coronary Syndrome in a Population-Based Nested Case-Control Study. J. Pers. Med. 2022, 12, 110. https://doi.org/10.3390/jpm12010110
Malyutina S, Chervova O, Tillmann T, Maximov V, Ryabikov A, Gafarov V, Hubacek JA, Pikhart H, Beck S, Bobak M. The Relationship between Epigenetic Age and Myocardial Infarction/Acute Coronary Syndrome in a Population-Based Nested Case-Control Study. Journal of Personalized Medicine. 2022; 12(1):110. https://doi.org/10.3390/jpm12010110
Chicago/Turabian StyleMalyutina, Sofia, Olga Chervova, Taavi Tillmann, Vladimir Maximov, Andrew Ryabikov, Valery Gafarov, Jaroslav A. Hubacek, Hynek Pikhart, Stephan Beck, and Martin Bobak. 2022. "The Relationship between Epigenetic Age and Myocardial Infarction/Acute Coronary Syndrome in a Population-Based Nested Case-Control Study" Journal of Personalized Medicine 12, no. 1: 110. https://doi.org/10.3390/jpm12010110