1. Introduction
Lung cancer is one of the deadliest and most common cancer types, accounting for 2.2 million cases and 1.8 million deaths worldwide in 2020 [
1]. Non-small-cell lung cancer (NSCLC) is the most prevalent sub-type of lung cancer (LC), representing around 80–85% of the cases [
2]. Depending on various factors, two types can be differentiated within NSCLC: lung adenocardinoma (LUAD) and lung squamous cell carcinoma (LUSC). LUAD can be found in the peripheral lung tissue [
3], while LUSC is usually centrally located [
4,
5]. An appropriate identification of the NSCLC lung cancer subtype is critical in the diagnostic process, since therapies differ for LUAD and LUSC [
6]. With the advances of computational methods, clinical decision support systems (CDSSs) have been created for cancer detection using biological sources, achieving great results. Among the data sources used in the literature, we have found, for instance, whole-slide imaging (WSI) [
7], gene expression data [
8], copy number variation (CNV) analysis [
9], miRNA expression data [
10], or DNA methylation (metDNA) values [
11]. By using these modalities independently, an accurate diagnosis can be performed. However, in their inner nature, they provide different biological information that may complement or regulate the information provided by the others. For instance, studies have shown that miRNAs regulate specific genes related to the proliferation of NSCLC [
12,
13] or methylation and mutation patterns have been predicted using WSI [
7,
14]. Therefore, exploring whether the fusion of them can provide a more robust diagnosis using computational methods is of great interest for improving the prognosis of the patient.
Information fusion has been a topic of interest in machine learning (ML) in the last decades given the immense amount of heterogeneous information that is being gathered in problems from all areas. The main premise of these methodologies is that the fusion of the information provided by different sources can achieve better results than those obtained by independent classifiers. Three different approaches can be distinguished depending on when the fusion takes place: late, early, and intermediate fusion [
15,
16,
17]. In the late fusion independent classifiers, one for each source of information is trained over the available training data. Then, the outputs produced by these classifiers are fused in order to provide a final prediction, for instance using a weighted sum of the probabilities or by using a majority-voting scheme [
18]. By doing so, the mistakes performed by some classifiers can be compensated by the others, improving the final classification. In addition, using a late fusion strategy allows dealing with missing information, which is a very typical setting in biomedical problems.
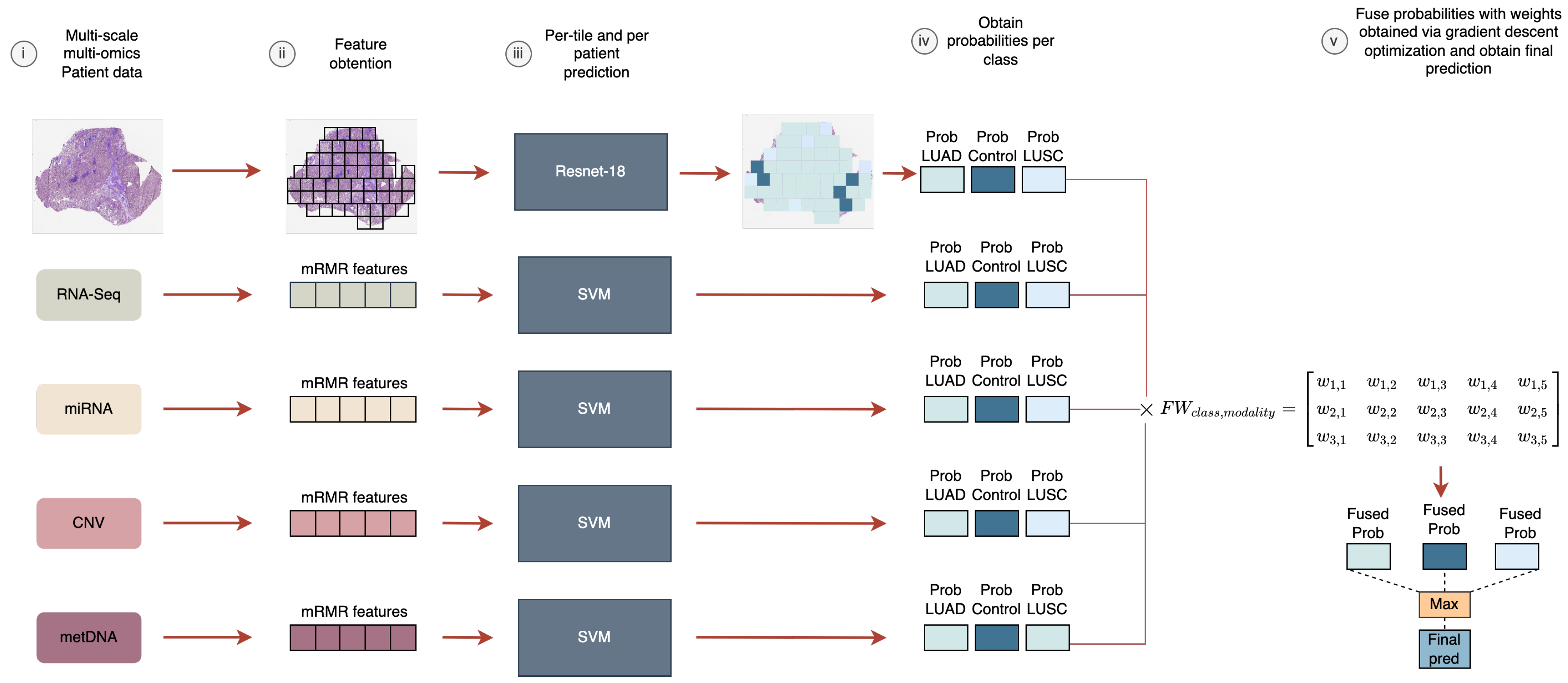
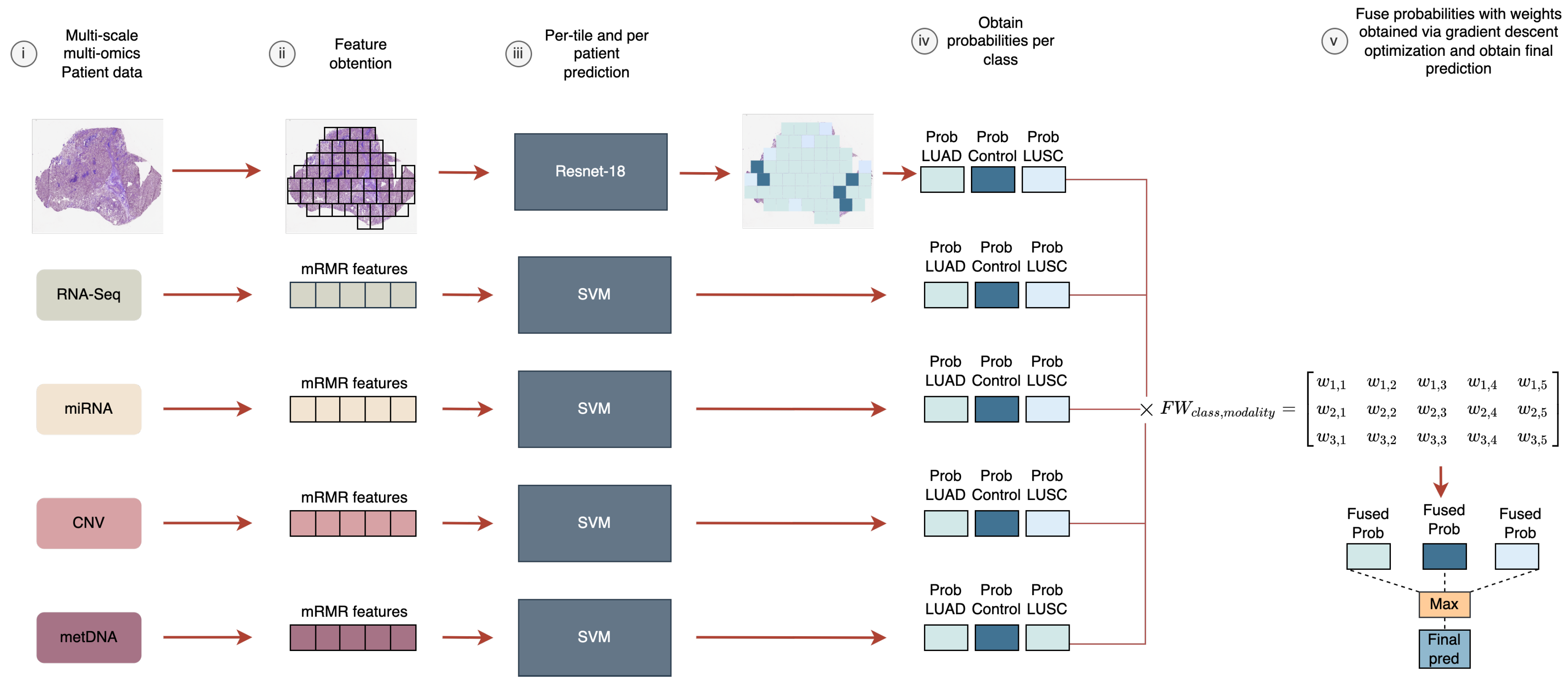
In this work, we aimed to analyze the fusion of five heterogeneous modalities (WSIs, RNA-Seq, miRNA-Seq, CNV, and metDNA) using a late fusion approach for the LUAD vs. LUSC vs. control classification problem. We evaluated the improvements that can be obtained by fusing information, and the modalities that are crucial to differentiate between the sub-types. In addition, a new late fusion optimization methodology is proposed for this problem, where the weights for the weighted sum of the probabilities are obtained by using a gradient descent approach that takes into account the performance of the fusion model in the classification.
2. Related Work
Over the last few years, the potential of ML models using biological data for the diagnosis and prognosis of cancer patients has been shown. Specifically, all the aforementioned biological sources have been used for the creation of CDSS in lung-cancer-related problems.
The use of gene expression data for lung cancer type classification has been explored in the literature in recent years, especially for LUAD given that it is the most frequent NSCLC type. Smolander et al. reached 95.97% accuracy in the LUAD vs. control problem using coding RNA and employing a deep learning model [
19]. Likewise, Fan et al. approached the same problem but used support vector machines (SVMs) with a 12-gene signature, obtaining an accuracy of 91% [
20]. In addition, some works have been presented for the multiclass classification of lung cancer subtypes. Gonzales et al. presented a model for the classification of small-cell lung cancer (SCLC), LUAD, LUSC, and large-cell lung carcinoma (LCLC) by finding differentially expressed genes (DEGs) and using them as input [
21]. By employing RF as the feature selector and k-NN as the classification algorithm they obtained an accuracy value of 88.23%. Castillo-Secilla et al. reached an accuracy of 95.7% using the random forest algorithm in the NSCLC subtype classification task [
22]. For the case of miRNA-Seq analysis, some works have been presented in the literature for lung cancer classification. Ye et al. presented a 10-miRNA signature for LUSC vs. control classification, reaching an F1 score of 99.4% [
10]. Yang et al. presented an miRNA signature for pathological grading in LUAD [
23], reaching an accuracy of 66.19%. In addition, miRNA has shown its potential for pancancer prognosis and treatment recommendation, including LUSC [
24]. CNV data have also been used in the literature for lung cancer classification. Qiu et al. presented a CNV signature for LUAD, LUSC, and control classification formed by 33 genes reaching an accuracy of 84% in the validation set [
9]. metDNA data have been used in the literature for LUAD vs. control classification, reaching an accuracy of 95.57% by Shen et al. [
25]. In addition, the relation of DNA methylation-driven genes with LUSC and LUAD classes was studied by Gevaert et al., finding the clusters of methylation-driven genes that provided clinical implications [
26]. Cai et al. tested different feature selection algorithms in combination with different ML algorithms for the task of LUAD vs. LUSC vs. SCLC classification, reaching an accuracy of 86.54% on the task by using a panel of 16 CpGs sites [
11].
Deep learning (DL) has shown great potential for computer vision tasks, and therefore, its use combined with WSI has been explored in the literature for NSCLC subtype classification. Coudray et al. presented a convolutional neural network (CNN) using tiles extracted from WSI for LUAD vs. LUSC vs. control classification and mutation prediction, finally reaching an area under the curve (AUC) score of 0.978 in the classification task [
7]. By using images manually labeled by experts, Kanavati et al. presented a CNN model using transfer learning for the lung carcinoma vs. control problem, and obtained an AUC score of 0.988 [
27]. Finally, other approaches have been presented where deep learning has been combined with more traditional statistics. Graham et al. used tiles extracted from the images and summary statistics to perform the classification between LUAD, control, and LUSC, reaching an accuracy value of 81% [
28].
The fusion of the aforementioned sources has been explored in the literature for various lung cancer problems, such as prognosis, grading prediction, or analyzing the relation between them. A deep neural network (DNN) was developed by Lai et al. that combined gene expression and clinical data for prognosis prediction in NSCLC patients [
29]. More novel techniques, such as autoencoders, have been explored in the literature for the generation of a feature representation for a later fusion. Cheerla et al. used a deep-learning-based model using miRNA, RNA-Seq, clinical, and WSI data for a pancancer prognosis prediction problem [
17]. Similarly, Lee et al. used an autoencoder for obtaining feature representation using mRNA, miRNA, CNV, and metDNA for prognosis prediction [
30]. For the problem of grading prediction, Long et al. proposed to use a late fusion methodology along with a gcForest model for predicting the stage of LUAD by fusion RNA-Seq, metDNA, and CNV [
31]. The authors reached an F1 score of 88.9% on the task. Finally, in a previous work we showed that the fusion of WSI with RNA-Seq data improved the results obtained by each independent source for the LUAD vs. LUSC vs. control problem [
32].
As detailed, previous research has focused on the use of single modalities for the classification, obtaining great results with both molecular and imaging approaches. However, fewer works have been presented in the literature performing a fusion of the information provided by these modalities, missing the opportunity to improve the classification performance and the knowledge acquisition from multiple biological sources. We propose to use the multimodal information to enhance the classification performance for the subtype identification, by leveraging the performance of independent classifiers and exploring the improvements that each source provides. A summary of the different works described for NSCLC classification problems is presented in
Table 1.
4. Results and Discussion
4.1. Performance of Each Data Modality
For the late fusion strategy we need to train independent models using each data modality. In the case of the molecular data, the number of features for each modality was selected based on having the lower number of features that provided the best performance for each independent model, by using the training splits in the 10-fold CV process. Finally, 6 genes were selected for RNA-Seq, 9 miRNA for miRNA-Seq, 12 genes for CNV, and 6 CpGs sites for metDNA data.
The results that were obtained when using each source of information separately can be observed in
Table 4, using all the available samples for the three-class classification problem (see
Table 2). For the independent models, the higher results for the classification are obtained when using RNA-Seq and metDNA, followed by miRNA-Seq (see
Table 4). These results are in accordance with previous studies in NSCLC. Qiu et al. obtained an accuracy of 84% for CNV data [
9]. Similarly, the results obtained by Cai et al. (an accuracy of 86.54%) using metDNA are improved by those we have obtained [
11]. For the case of WSI, the presented results are very similar to those obtained by Coudray et al. (an AUC of 0.978) and Graham et al. (an accuracy of 81%) [
7,
28]. For RNA-Seq, Castillo-Secilla et al. reached an accuracy of 94.7% using SVMs, which is similar to our obtained performance [
22].
4.2. Performance of Late Fusion with Different Number of Sources
Once the models were trained, we tested the different improvements that can be obtained when adding new information, comparing the fusion of the sources in groups of two, three, four, and five. By doing so we were able to see how sources complemented each other in terms of classification performance, and when they improved or worsened it. These results can be observed in
Table 4. For the late fusion models we used those samples that the data modalities in use have in common. The number of samples per class are provided as
Supplementary Material (see Tables S1 and S2). The confusion matrices for the discussed fusion models are provided as
Supplementary Material (see Figure S1).
When fusing two sources, the highest performance in terms of classification metrics was obtained for the fusion of WSI-RNA-Seq, RNA-Seq-miRNA, and RNA-metDNA. Given that RNA-Seq, miRNA-Seq, and metDNA were the ones that achieved the highest performance independently, it was expected that their fusion would provide an increase in the metrics. However, the fusion of WSI and RNA-Seq achieved great results in the classification, even though WSI was not among the sources with the best independent metrics. Therefore, WSI must be improving some of the RNA-Seq predictions, which might be on the wrong side of the prediction border of the probabilities.
Then, we moved to using three sources for the late fusion model. By adding miRNA data to the WSI-RNA-Seq fusion model the results obtained improved (from to in terms of F1 score). The same happened when we included CNV or metDNA in the RNA-Seq-miRNA fusion model. RNA-Seq seems to be the most important source, since it was included in those fusion models with a high performance. In addition, the fusion of RNA-miRNA with other sources improved the classification over using RNA-Seq independently or with other sources.
Finally, we carried out experiments to observe whether there was an improvement in the classification performance when using four or five sources. In this case, the only fusion that improved results over the fusion of three sources, in terms of the F1 score and very similar results in the accuracy metric, was when we fused all the biological sources. However, the improvement was really small and the standard deviation increased (
for WSI-RNA-Seq-miRNA and
for the fusion of all sources). For the rest of the fusion cases, the results obtained are similar to the highest reached when using three sources of information (see
Table 4). Therefore, performing more screenings for the patient if you already have the biological sources that provided the best performance when using three sources is not necessary for an accurate diagnosis in this case.
4.3. Performance of the Fusion Models with Missing Information
Dealing with missing information is crucial when working with biological sources, given the high cost of some of the screenings. Therefore, we evaluated the effectiveness of the fusion model when some of the modalities were missing. In order to do so, for each fusion model the metrics were computed on all the samples available for the fused modalities, without restricting to those that the modalities have in common. In
Figure 2, the receiver operating characteristic (ROC) curves obtained by the fusion model with all modalities can be observed. In
Figure 3, the F1 score is presented for each fusion case predicting on all the samples that each modality has (see
Table 2).
Except for miRNA-Seq samples, the fusion that achieves the best performance is when fusing the five sources. However, the improvement is small in comparison with using four sources, so not having all of them does not excessively affect the classification performance. Fusing only CNV with metDNA RNA or miRNA-Seq worsens the performance in comparison to the usage of them independently, which could be due to the imbalance in the classes (
Table 2). The combination of metDNA and WSI also performs poorly, maybe due to the fact that it has been shown in the literature that WSI reflects information about the methylation patterns of human tumors [
14], and therefore, they might not be complementing each other. However, in most cases, including additional information improves the results that can be obtained by each independent source.
The final results obtained when fusing all the data sources is an F1 score of
, an accuracy of
, an AUC of
, and an AUPRC of
. These results improved those aforementioned and also reduced the standard deviation obtained across the splits. The ROC curves (
Figure 2) obtained show the performance of each individual modality and the fusion model over all the available samples for each one (all the samples in the case of the fusion model). The fusion model outperforms each modality for the three classes, showing the potential of using all the information. In addition, the fusion model reduces the number of misclassified samples for all sources, representing a reduction in the diagnosis error rate rate up to ≈8.6% in the best case and ≈1.6% in the worst case (see
Table 5). The confusion matrix obtained over the whole dataset is presented in the
Supplementary Material (see Figure S2) along with the weights obtained for each modality in the fusion (see
Supplementary Material Table S4).
4.4. Comparison with Previous Work
The majority of the works presented in the literature for NSCLC subtypes and control classification have focused on using a single data modality and mainly a bi-class classification problem. Our fusion model outperforms or reaches the same results obtained by those works where an LUAD vs. LUSC vs. control classification has been presented, and a summary is presented in
Table 6. The fusion of information improves the results that Qiu et al. obtained for CNV data (an accuracy of 84%) [
9]. Similarly, the results obtained by Cai et al. using metDNA are also improved (they obtained an accuracy of 86.54%) while reducing the number of CpG site signatures [
11] and similar results were obtained compared to those presented by Castillo-Secilla et al. using RNA-Seq (accuracy of 95.7%) [
22]. For the case of WSI, the fusion also improved the results presented in the literature by Coudray et al. [
7] (an AUC of 0.978) reaching an AUC of 0.991. In the case of multi-omic fusion, we have not found works presenting methods for the NSCLC subtypes and control classification. However, in other NSCLC-related problems the fusion of information has presented an enhancement in the performance. Cheerla et al. [
17] showed that by fusing clinical, miRNA, and WSI data, the performance was improved in LUAD prognosis prediction. Similarly, Lee et al. [
30] improved the prognosis prediction by fusing the information of four sources (RNA-Seq, miRNA, CNV, and metDNA) over each independent one. This same behavior was observed in our case for these sources.
When it comes to the relations between data modalities, our results highlight previously reported patterns. It has been presented in the literature that WSI can be used to predict mutation patterns or gene expression levels [
7,
51], so the information provided may be completed with the one presented in RNA-Seq data. Similarly, when fusing three data modalities it was shown that including miRNA-Seq in the RNA-Seq-WSI fusion model improved the classification performance. miRNAs regulate specific genes related to the proliferation of NSCLC [
12,
13], and therefore, might be complementing the information provided by RNA-Seq and WSI.
5. Conclusions
In this paper, we demonstrated the usefulness of fusing heterogeneous sources of biological information for NSCLC subtypes and control classification. In addition, we proposed a new optimization methodology for weighting the classifiers in a late fusion strategy, effectively dealing with missing information and reaching good performance in the classification.
The fusion of the information outperformed the use of each independent source for the classification. Independently, RNA-Seq and metDNA achieved the highest performance in the classification. When performing the fusion, RNA-Seq is crucial for the classification problem and the addition of miRNA-Seq in combination with another data modality improved the obtained results. The best results were obtained when fusing the five sources of information reaching an F1 score of when classifying all the available samples from all sources. However, there was not a huge increase in comparison with using three or four sources. The obtained results also highlight other reported patterns in the literature between data modalities that should be further studied. In addition, the methodology effectively deals with missing information, which is mandatory given that not all screenings are always performed to a patient. The presented methodology can be used in any diagnosing problem where heterogeneous sources of information are available, and it can be extended to any number of data sources.
As future work we would like to test the generalization capabilities of the proposed methodology for the classification of other cancer types or in other diagnosis-related problems and evaluate whether the relations found between the different modalities apply to these other problems.


 ,
, 



{kind=link}
{kind=link}
{kind=link}