Abstract
Background: The intestinal wound healing process is a complex event of three overlapping phases: exudative, proliferative, and remodeling. Although some mechanisms have been extensively described, the intestinal healing process is still not fully understood. There are some similarities but also some differences compared to other tissues. The aim of this systematic review was to summarize all studies with knockout (KO) experimental models in bowel anastomoses, underline any recent knowledge, and clarify further the cellular and molecular mechanisms of the intestinal healing process. A systematic review protocol was performed. Materials and methods: Medline, EMBASE, and Scopus were comprehensively searched. Results: a total of eight studies were included. The silenced genes included interleukin-10, the four-and-one-half LIM domain-containing protein 2 (FHL2), cyclooxygenase-2 (COX-2), annexin A1 (ANXA-1), thrombin-activatable fibrinolysis inhibitor (TAFI), and heparin-binding epidermal growth factor (HB-EGF) gene. Surgically, an end-to-end bowel anastomosis was performed in the majority of the studies. Increased inflammatory cell infiltration in the anastomotic site was found in IL-10-, annexin-A1-, and TAFI-deficient mice compared to controls. COX-1 deficiency showed decreased angiogenesis at the anastomotic site. Administration of prostaglandin E2 in COX-2-deficient mice partially improved anastomotic leak rates, while treatment of ANXA1 KO mice with Ac2-26 nanoparticles reduced colitis activity and increased weight recovery following surgery. Conclusions: our findings provide new insights into improving intestinal wound healing by amplifying the aforementioned genes using appropriate gene therapies. Further research is required to clarify further the cellular and micromolecular mechanisms of intestinal healing.
1. Introduction
Anastomosis construction is considered one of the most crucial steps in gastrointestinal (GI) surgery. An anastomotic leak is a potentially fatal complication, resulting in higher reoperation rates as well worse prognosis, with increased morbidity and mortality [1,2,3]. Furthermore, apart from the physical and psychological burden, an anastomotic leak represents a significant economic burden for healthcare facilities, mainly due to prolonged hospital stay [4]. Despite progress in GI surgery techniques and materials, GI anastomoses currently present high leakage rates; up to 4% for ileocolic, 18% for colorectal, and 19% for coloanal anastomoses [5]. The problem is exacerbated in patients with inflammatory diseases, sepsis, malnutrition, or in those receiving steroids, immunosuppressives, and neoadjuvant radio-chemotherapy [6].
There are three phases of intestinal wound healing, including inflammation, proliferation, and remodeling, which partly occur simultaneously. The inflammatory phase begins immediately after injury, in which leukocytes infiltrate the submucosa along with accompanying oedema. This process lasts for two weeks, initially with a neutrophil-rich acute response which slowly changes into a chronic macrophage response. Fibroblast and smooth muscle cell proliferation lead to small amounts of collagen formation in order to initially strengthen the anastomosis. Mucosal resurfacing starts early and is completed within one week. Remodeling starts after one week through additional collagen formation and substitution of the already existing collagen with stronger types of collagen. However, the architecture of muscularis mucosa and muscularis propria remains irregular compared to healthy tissue [7].
Intestinal anastomosis healing follows a process quite similar to that encountered in other tissues, with some remarkable differences. The rate of healing in the GI tract is rapid (weeks), whereas in the skin it is more prolonged (months) [8]. The serosa in the GI tract give additional healing strength. The shear stress in the GI tract is increased due to intraluminal bulk transit and peristalsis. GI healing bears a high aerobic and anaerobic bacterial load while skin only has aerobic bacteria. A study from Wahl et al. showed that E. coli species’ predomination in the GI tract can produce toxins, disrupting epithelial integrity and collagen synthesis [9]. Another difference exists in the cellular source of collagen; fibroblasts are responsible for this in the skin, whereas smooth muscle cells are responsible for this in the GI tissues [10]. Furthermore, there are differences in the healing process between different sections of the GI tract. For example, collagen synthesis occurs more rapidly in the ileum compared to the colon [8]. Small bowel anastomosis strength approaches the strength of unwounded tissue about four weeks post injury, while the large bowel needs four months to gain only 75% of its initial strength.
Despite our knowledge of GI healing processes, there are still many molecules and molecule interactions to be identified in order to get a complete idea of their importance and their participation in serious complications, such as improper healing leading to anastomotic leak. A useful tool to explore and analyze the action of certain molecules is the use of knockout mice. Knockout (KO) mice are genetically modified laboratory animals lacking a certain gene and its codified protein. KO mice have been increasingly utilized in recent years to study the immunological and molecular mechanisms involved in anastomotic would healing and interactions therein. This systematic review aims to summarize all significant findings of KO mice experiments in GI wound healing and potentially clarify their relationship to anastomotic leak occurrence.
2. Materials and Methods
2.1. Study Design and Inclusion/Exclusion Criteria
This systematic review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, in line with the protocol developed and agreed a priori by all authors. Studies investigating the impact of genetic KO mouse or rat models’ effects on bowel anastomosis integrity were deemed eligible for analysis. Control mice or rats among the studies were animals that received no intervention, drinking normal water, or being mentioned in the text as wild-type animals. Exclusion criteria were as follows: (i) articles published in languages other than English, (ii) narrative or systematic reviews and meta-analyses, (iii) case reports, errata, comments, perspectives, letters to the editor, and editorials that did not provide any extractable data, (iv) published abstracts with no available full text, and (v) non-comparative studies (single-arm studies). No publication date, sample size restrictions, or any other search filters were applied.
2.2. Search Strategy
Eligible studies were identified by searching through the MEDLINE (via PubMed), Cochrane Library, Embase, and Scopus databases (end-of-search date: 1 October 2023). These searches were conducted by two independent researchers. The search strategies used were the following: (knockout) AND (anastom*). The search algorithm included all of the available synonyms and related terms. Any disagreements were resolved by a third reviewer. The reference lists and all previously published systematic reviews were thoroughly searched for missed studies eligible for inclusion based on the “snowball” methodology [11].
2.3. Data Extraction
A standardized, pre-piloted form was used for data tabulation and extraction. Two reviewers extracted the data independently, and any disagreements were identified and resolved by a third reviewer. We extracted the following data from the included studies: study characteristics (first author, year of publication, country, and number of mice in each group), (ii) gene characteristics (KO gene, gene function, underlying condition), (iii) operation characteristics (method of anastomosis, suture material), and (iv) post-operative analysis (hydroxyproline, bursting pressure, histological characteristics).
3. Results
3.1. Study Characteristics
A total of one hundred and forty studies were identified and a total of eight studies were included for data extraction, published between 2003 and 2021 [12,13,14,15,16,17,18,19]. Two studies were from Germany [15,16], two studies were from the USA [12,13], another two were from the Netherlands [14,18], one was from Canada [19], and one from China [17]. All studies included KO mouse models. A total of 767 mice were included in the review, averaging 95.9 mice per study (Table 1). A flow diagram is shown in Figure 1.

Table 1.
Summary of the study details and genetic knockout characteristics. ANXA1 = annexin A1, COX-2 = cyclooxygenase-2, HB-EGF = heparin-binding epidermal growth factor, IL-10 = interleukin-10, KO = knockout, MAPK = mitogen-activated protein kinase, NA = not applicable, NSAID = non-steroidal anti-inflammatory drug, SHIP1 = Src homology-2 domain-containing inositol 5-phosphatase 1, TAFI = thrombin-activatable fibrinolysis inhibitor, TG = transgenic, VEGF = vascular endothelial growth factor, WT = wild type (act as control for the genetically altered mice).
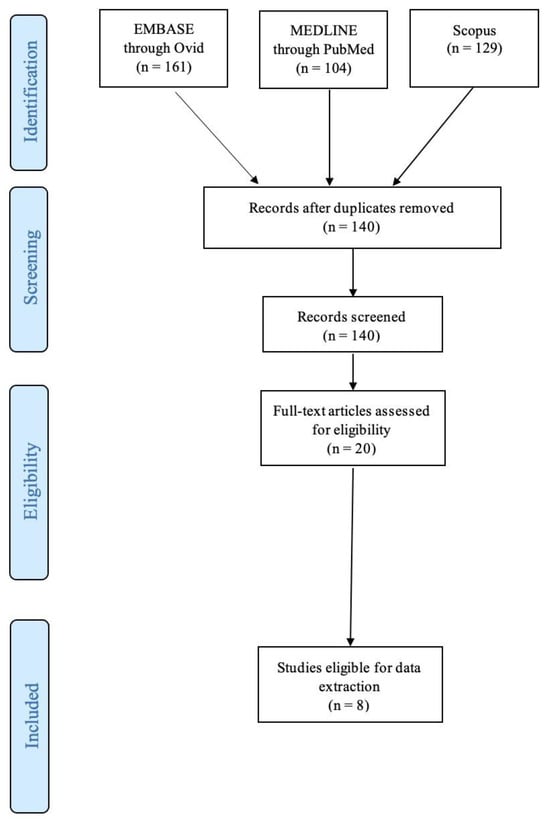
Figure 1.
PRISMA flowchart.
3.2. Experimental Animals Characteristics
Three studies used IL-10 KO mice [12,17,19]. In one of these studies, triptolide was used to reverse the pro-inflammatory effect of IL-10 deficiency [17]. These mice subsequently developed Crohn’s disease as a result of IL-10 deficiency. One study modeled Crohn’s disease by FHL2 deletion, a multifactorial cofactor for gene transcription which negatively regulates mitogen-activated protein kinase (MAPK) signaling [16]. Another study used cyclooxygenase-2 (COX-2) KO mice mimicking a non-steroidal anti-inflammatory drug (NSAID) effect. The result was immune modulation, myoblast proliferation, and reduced vascular-endothelial-growth-factor- (VEGF-) induced angiogenesis [14]. One study used annexin A1 (Anxa1) gene KO mice to induce inflammatory colitis by leukocyte activation and transmigration [15]. Another study used thrombin-activatable fibrinolysis inhibitor (TAFI) KO mice, compared to wild type (WT) mouse models, for anastomotic studies [18]. One study used heparin-binding epidermal growth factor (HB-EGF) KO mice for their experiments, again compared to WT mice used as control [13] (Table 1).
3.3. Operative Characteristics
The majority of studies used interrupted sutures to form an end-to-end anastomosis [12,13,14,15,16,17]. One study used continuous sutures for their end-to-end anastomosis [18] and another study formed a side-to-side anastomosis with interrupted sutures [19]. Three studies used 8-0 prolene for the anastomosis [14,18,19]. Two used 9-0 nylon [12,17], and one used 4-0 nylon [13]. Two studies used vicryl sutures [15,16] (Table 2).
Table 2.
Summary of operative details, histological and inflammatory analysis, and bursting pressure. ANXA1 = annexin A1, COX-2 = cyclooxygenase-2, HB-EGF = heparin-binding epidermal growth factor, IL-10 = interleukin-10, KO = knockout, MMP = matrix metalloproteinase, NA = not assessed, POD = postoperative day, TAFI = thrombin-activatable fibrinolysis inhibitor, TG = transgenic, VEGF = vascular endothelial growth factor, WT = wild type.
3.4. Histological Characteristics
All studies assessed the histological characteristics of the bowel anastomosis in gene KO mice. In IL-10 KO mice, a study found no significant difference in collagen deposition between IL-10 KO mice and control [19], while another study found significantly increased collagen mRNA in IL-10 KO mice compared to WT mice. This additional collagen mRNA translated into increased collagen deposition and fibrosis at the anastomosis [12]. The microbiome is also likely to play a role, with reduced fibrosis in germ-free IL-10 KO mice compared to conventional IL-10 KO mice [12], although the exact mechanisms have not been identified. Wu et al. [17] found epithelial cell hyperplasia, mucin depletion, and crypt abscess formation in IL-10 deficient mice at the anastomosis site and more extensively throughout the bowel.
FHL2 KO mice demonstrated reduced submucosal collagen type I and III deposition around the injury site, with no difference in collagen in areas of unaffected bowel compared to WT mice acting as controls [16]. Inhibition of COX-2 correlated with reduced vascularization around the anastomosis site compared to controls [14]. Annexin A1 KO mice demonstrated a reduced histological healing score (Scored 0–4 for inflammatory cells, angiogenesis, collagen synthesis, fibroblast ingrowth, and overall healing quality, adapted from Phillips et al. [20]) compared to control, which was improved with the addition of Ac2-26 nanoparticles [15]. TAFI KO mice showed no differences in granulation tissue deposition between the KO group and WT [18]. HB-EGF KO mice showed reduced healing scores compared to WT. Transgenic (TG) HB-EGF mice, engineered to overexpress HB-EGF resulting in additional HB-EGF produced, showed increased collagen deposition and mature vascularization around the anastomosis site compared to WT mice [13]. Seven studies assessed inflammation differences around the anastomosis site [12,13,14,15,17,18,19]. IL-10 KO mouse models showed increased lymphocyte infiltration when compared to WT mice that faced either surgical intervention or a non-operative study arm [12,17,19]. This was reversed with administration of triptolide [17]. Increased inflammation (defined as increased numbers of morphonuclear leukocytes, lymphocytes, and macrophages on histological analysis) was found in HB-EGF KO, compared to WT, which demonstrated increased inflammation compared to TG HB-EGD mice [13]. Increased inflammation and matrix metalloproteinase (MMP) expression was found in Anxa1 KO mice compared to WT, with reduced inflammation upon addition of Ac2-26 nanoparticles [15]. However, no anastomotic area inflammatory differences were noted between COX-2 KO mice and WT mice [14]. Interestingly, Te Velde et al. [18] only found increased inflammation around the anastomotic site in TAFI KO mice who experienced anastomotic leak, but not in other TAFI KO mice (Table 2).
3.5. Bursting Pressure
Three studies assessed bursting pressure. Kirfel et al. [16] demonstrated a reduced bursting pressure in FHL2 KO mice compared to control at days 2, 5, and 14, postoperatively. TAFI KO mice also had a reduced bursting pressure despite no granulation tissue differences when compared to WT mice [18]. HB-EDF KO mice had reduced bursting pressure compared to WT mice. TG HB-EGF mice had increased bursting pressure when compared to WT, again correlating with collagen deposition noted on histological analysis [13] (Table 2).
4. Discussion
Bowel resection and anastomosis represents a significant component of GI disease surgical management. Anastomotic leak represents a severe post-operative complication, with varying incidence up to 36% [21]. The clinical presentation of leaks varies significantly, from low-grade fever and overall delayed recovery of the patient to severe peritonitis and sepsis [22]. This results in longer inpatient stays, increased mortality, and higher healthcare expenses [23,24,25]. Therefore, it is of utmost importance to elucidate intestinal wound healing pathways in order to find ways to improve healing and reduce the incidence of anastomotic leaks. This systematic review identified several studies investigating the lack of the following molecules: IL-10, cyclooxygenase-2, annexin A1, TAFI, and heparin-binding epidermal growth factor genes. Increased inflammatory cell infiltration in the anastomotic site was found in IL-10-, annexin-A1-, and TAFI-deficient mice compared to controls. COX-1 deficiency showed decreased angiogenesis at the anastomotic site. Our findings provide new ways to improve intestinal wound healing through amplifying the aforementioned genes with appropriate gene therapies. Genetic modification of mouse models has translated into different results of clinical parameters. Various agents have been shown to be useful in reducing anastomotic leaks and their relative complications. For example, Radulescu et al. [13] generated mouse models over-expressing HB-EGF. This increased bursting pressure and collagen deposition. In a different included study conducted by Reischl et al., Ac2-26 nanoparticles added to ANXA1 KO mice improved histological healing [15]. Addition of prostaglandin E2 in COX-2-deficient mice partially improved anastomotic leak rates, closer to those of WT mice [14]. HB-EGF KO mice models led to higher mortality and complication rates (including bleeding, anastomotic dehiscence, abscess formation, and obstruction) [13]. Treatment of ANXA1 KO mice with Ac2-26 nanoparticles reduced colitis activity and increased weight recovery following surgery [15]. Use of diclofenac to inhibit COX-2 resulted in significantly increased rates of anastomotic leak compared to control [14]. TAFI KO mice had significantly increased weight loss, adhesions, bleeding, and mortality compared to WT mice [18].
4.1. Interleukin 10, miR-155/SHIP-1 Pathway and Wound Healing
IL-10, as a regulatory cytokine of inflammation, has been the subject of extensive study in the context of anastomotic healing. IL-10 is primarily produced from macrophages residing in the lamina propria of the bowel and regulatory T-cells in order to reduce the immune response to proinflammatory molecules on cell membranes of local bacteria [26]. In addition, epithelial cells and Th1 cells triggered by lipoproteins of commensal bacteria are capable of producing IL-10 in order to maintain bowel homeostasis [27,28]. Incubation of IL-10 KO mice’s intestinal epithelial cells with recombinant IL-10 enhanced wound repair in vitro because IL-10 binding reversed the KO effect [29]. In vivo, macrophage-derived IL-10 was crucial for small intestine epithelial healing during the acute phase of injury, ultimately driving re-epithelialization in the small intestine [30]. The exact mechanism through which IL-10 promotes bowel wound healing remains unclear. Following biopsy-induced injury, IL-10 activates cyclic adenosine monophosphate (c-AMP) Response-Element-Binding Protein (CREB) signaling and its downstream target WNT1-inducible-signaling pathway protein 1 (WISP-1). This, in turn, promotes β-catenin signaling to induce epithelial proliferation and wound closure [31]. It is therefore possible that IL-10 works directly on epithelial cells. An alternative mechanism suggests that IL-10 works primarily on immune cells, resulting indirectly in wound healing. IL-10 binding to CD11+ myeloid cells regulates T cell activation and proliferation of intestinal crypt cells [12,32]. The broad functions of IL-10 have generated significant interest in it as a therapeutic target for multiple disease states. Significant research involving manipulation of IL-10 has been undertaken in the context of inflammatory bowel disease (IBD) [25]. IL-10-deficient mice develop colitis [33], which is managed successfully following administration of IL-10 [34,35]. In 1993, Kühn et al. [36] showed that interleukin-10-deficient (IL-10−/−) mice presented a Th1-mediated chronic colitis similar to human IBD. However, administration of recombinant IL-10 yielded only a minor clinical improvement in the treatment of IBD in humans [37], likely due to the low concentrations achieved in the gut following systemic administration. Lactococcus lactis bacteria, modified to deliver IL-10 to the gut, improved clinical disease scores of a small number of Crohn’s disease patients [38]. The effects of IL-10 have also been studied in dermal wound healing. Fetal skin has a tendency to regenerate, whereas postnatal skin heals with scar formation. Fetal skin has elevated levels of IL-10, promoting high molecular weight hyaluronan formation, which reduces inflammation and fibrosis [39,40]. IL-10 administration in IL-10-deficient mice resulted in reduced inflammatory cell numbers and restoration of normal skin architecture, with reduced scar size. This resulted in accelerated dermal healing and increased tissue strength [41]. Regarding IBD, it has been demonstrated that triptolide ameliorates the severity of IL-10-deficient mouse colitis via inhibition of TLRs (toll-like receptors)/NF-κB and IL-6/STAT3 signaling pathways, and down-regulation of IL-17 [42,43]. Triptolide is a bioactive compound produced by the plant Tripterygium wilfordii Hook F (TwHF) and has been widely used in clinical practice due to its anti-inflammatory and immunosuppressive properties [44]. Its mechanisms of action include the following: (1) inhibition of expression of RPB1 (the RNA polymerase II main subunit), (2) inhibition of xeroderma pigmentosum group B (XPB) protein, which is a subunit of transcription factor II-H, and (3) inhibition of induction of miR-155 in LPS-stimulated macrophages [17]. In particular, miR-155 targets SHIP-1 (Src-homology-2-containing inositol phosphatase-1), which is an inhibitor of many inflammatory processes and regulates the balance of T cell subsets [17,42,43].
4.2. Cyclooxygenase (COX) and Wound Healing
COX is an essential enzyme in the synthesis of prostaglandins, occurring in two isoforms. COX1 is constitutively expressed, whereas COX2 is expressed in response to injury. In dermal injury, COX-2 expression peaks 3 days later, when the anastomosis is thinnest and at increased risk of rupture [45]. The quantity of COX-2 produced also correlates with the magnitude of the inflammatory response [46]. Hypoxia, partly due to connective tissue deposition during wound healing, stimulates angiogenesis through HIF-1α induction of VEGF secretion. VEGF is also responsible for fibroblast differentiation and tissue remodeling. The COX–VEGF axis plays an important role in wound healing, since pharmacologically induced downregulation of COX-2 correlates with significantly increased VEGF levels. A study of full-thickness skin incisions in mice demonstrated increased angiogenesis and collagen deposition on histology, which correlated with a significant increase in wound contraction and reduced wound area macroscopically [47]. These findings are in contrast to those of another paper included in our analysis, indicating that COX-2 KO mice demonstrate reduced VEGF-induced angiogenesis at the anastomosis [14]. This could be due to an alternative mechanism for VEGF production, or a difference in healing between dermal and bowel injury. However, COX-2 and the resulting production of prostaglandin E2 have been shown to accelerate healing of gastric ulcers, further demonstrating the importance of this pathway in gastrointestinal mucosal repair [48]. Prostaglandin (PG) biosynthesis is mediated by the synergic action of two enzymes: phospholipase, responsible for the metabolism of arachidonic acid (AA) from cell membrane phospholipids, and COX. Prostaglandin E2 (PGE2) is the most abundant and important COX-2 product regarding intestinal wound healing [49]. Under normal conditions, the PGE2 signaling pathway can potentially trigger stem cell differentiation towards enterocytes [50]. After an injury, epithelial restitution is a crucial process that is performed in the intestine by a special cell population with transient repair features named wound-associated epithelial cells (WAE) and, in combination with non-canonical Wnt signaling activation, this ensures controlled wound repair [51]. Epithelial restitution is a protective mechanism including the migration of epithelial cells from intact wound margin into the injured basal lamina to reseal the intestinal barrier. Miyoshi et al. showed that PGE2 signaling through its receptor promotes WAE differentiation of intestinal epithelial stem cells in an in vitro model. In vivo studies in mice with depletion of the PGE2 receptor led to insufficient intestinal wound repair due to impaired production of WAE cells, and they exhibited impaired wound repair [52]. In addition, it has been found that increased COX-2 expression is noted in macrophages and myofibroblasts after exposure to proinflammatory signals, triggering intestinal tissue proliferation [14]. Numerous studies have shown the regenerative role of PGE2, a product of the COX-2 gene, for intestinal wall repair under a wide range of pathological circumstances, such as dextran-sulfate-sodium- (DSS-) induced inflammation and ischemia-reperfusion injury [53,54]. In addition, PGE2 and COX-2, through activation of the PGE2 receptor and following upregulation of VEGF expression, stimulate neoangiogenesis but also promote the proliferation and migration of epithelial intestinal cells [55]. Finally, PGE2 regulates myofibroblast function, which is responsible for producing collagen, the main structural component of the extracellular matrix [56]. In terms of clinical application, there is conflicting evidence regarding whether COX-2 blockage is associated with impaiastomotic leak development. Two recent meta-analyses involving patients undergoing operations for colorectal cancer have concluded in undetermined relationships between postoperative NSAID administration and anastomotic leakage [57,58]. On the other hand, a subgroup analysis by Jamjittrong et al. focused on colorectal anastomoses revealed a significantly increased anastomotic leakage rate in patients who were administered NSAIDs perioperatively [57]. In human genome studies, the PTGS2 −765G > C polymorphism lies in the promoter region of the gene that encodes COX-2 and leads to lower levels of COX-2 [59]. Reisinger and Makar suggested that the homozygous state for PTGS2-765G > C polymorphism, which is encountered with a prevalence of 3% in humans, led to decreased COX-2 expression and increased anastomotic leakage rates [14,60].
4.3. Anxa1 and Wound Healing
Annexin A1 (AnxA1), also named lipocortin-1, is a calcium-dependent phospholipid-binding protein from the family of SPMs. Clinical studies have shown that higher AnxA1 expression is associated with milder symptomatology and severity in both Crohn’s disease (CD) and ulcerative colitis (UC) [61]. A 26-amino acid ANXA1 N-terminal peptide Ac2-26, a part of the full-length molecule, is the active part of ANXA1 [62]. AnxA1’s anti-inflammatory role is based on inhibition of neutrophil accumulation, transendothelial migration, and activation, as well as promotion of neutrophil apoptosis after binding to the formyl peptide receptor 2 (FPR2/ALX) and formyl peptide receptor 1 (FPR1) [15]. In addition, AnxA1 induces monocyte chemotaxis and removal of apoptotic leukocytes by macrophages. It promotes keratinocytes’ motility and differentiation, which is important for wound re-epithelization [63]. Finally, AnxA1 controls macrophage cell reprogramming towards reduced production of proinflammatory molecules and increased production of anti-inflammatory agents [64]. Annexin 2 is a calcium-dependent phospholipid-binding protein associated with cytoskeleton coordination [65]. Through annexin 2 [66], epithelial cell migration re-establishes the epithelial barrier and reseals the mucosal defect of the bowel following surgery. Our review included a study [15] investigating the role of annexin A1 on the resolution of inflammation following surgery for colitis. Epithelial injury of the bowel brings into contact the immune cells of the intestinal wall and the lumen’s bacteria, resulting in an inflammatory response. Anxa1 is a component of the extracellular vesicles of these immune cells [67]. When released into the extracellular space, it modifies the inflammatory response of phagocytes and epithelial cells through binding to formyl peptide receptors [68]. Our included study [15] found an increased inflammatory score and increased MMP in Anxa1 KO mice, which is consistent with the process described above. Therefore, Anxa1 deficiency reduces epithelial integrity by exposing inflammatory cells to the gut microbiome. Clinically, increased Anxa1 expression in mouse models of CD correlated with histological recovery of the gut mucosa [69]. Further evidence suggests that Anxa1 mediates the positive response of infliximab in CD patients [70].
4.4. FHL2 and Wound Healing
FHL2 belongs to the LIM protein superfamily and exhibits a key role in a wide range of cell functions, including regulation of cell proliferation, gene expression, survival, control of cell architecture, cell adhesion, apoptosis, cell motility, and signal transduction [71]. FHL2 has a distinctive signaling role for mesenchymal cells, controlling their migration, contraction, and differentiation into myofibroblasts through expression of α-smooth muscle actin [16]. During the inflammation phase, FHL2 contributes to pathogen clearance, synthesis, and release of proinflammatory cytokines and immune cell infiltration. During the migration/proliferation phase it has a key role in the function of myofibroblasts, in their differentiation from fibroblasts or from epithelial cells via epithelial mesenchymal transition, and in suppression of MMPs, leading to sufficient contraction and formation of granulation tissue. Finally, during the remodeling phase of the wound healing, FHL2 participates actively in ECM synthesis and remodeling [72]. The literature contains many reports regarding the role of FHL2 in cardiovascular disorders and carcinogenesis [73,74]. However, emerging evidence indicates its role in all phases of wound healing and inflammation. Although under normal conditions FHL2 expression is very low in most tissues, it is upregulated in the skin during wound repair, especially in phases of migration and proliferation. FHL2 suppression is associated with healing failure or prolonged wound healing [72]. In terms of anastomosis healing, Kirfel et al. [16] explored the healing of an end-to-end small bowel anastomosis in FHL2-deficient mice compared to WT mice. They found that FHL2 KO mice presented a statistically significant increase in anastomotic failure rate and lower anastomotic bursting pressure. Histopathologically, insufficient wound healing was noted in anastomotic specimens derived from KO mice, characterized by decreased mucosal and muscular continuity and reduced granulation tissue formation at the fifth and fourteenth postoperative day. On the fifth postoperative day, WT mice presented a dense pattern of organized collagen formation, mainly in the submucosal layer of injured tissue, while submucosa in the FHL2 KO mice presented significantly thinner collagen I/collagen III networks. This was due to downregulation of collagen III mRNA expression in FHL2 KO mice at the anastomotic site. Depletion of FHL2 did not lead to difference in local MMP expression [16].
4.5. HB-EGF and Wound Healing
HB-EGF belongs to the epidermal growth factor (EGF) family. It binds to heparin, as well as to human epidermal growth factor receptors ErbB-1 and ErbB-4, and triggers cellular proliferation, migration, adhesion, and differentiation [75]. Especially for intestinal tissue, it has been shown that HB-EGF exerts a protective effect on intestinal epithelial cells, intestinal stem cells, immunocytes, vascular endothelial cells, pericytes, and intestinal neuronal cells after injury by preserving their cytoskeletal structure and proliferative potential [76,77]. Furthermore, its role in intestinal injury has been delineated in experimental models of necrotizing enterocolitis [78], ischemia/reperfusion injury [79], and resuscitation after hemorrhagic shock [80]. It has been demonstrated that HB-EGF repairs the intestinal epithelium via a phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase (ERK)1/2 pathways [81]. Angiogenesis and capillary formation are the basic mechanisms of action of the HB-EGF molecule, which has been shown both in vitro and in vivo [82]. To investigate the role of HB-EGF in intestinal anastomotic healing, Radulescu et al. [13] studied outcomes after division and re-anastomosis of the terminal ileum in HB-EGF KO mice and HB-EGF transgenic (TG) mice. In addition, an extra group of WT mice receiving per os HB-EGF (800 µg/kg) was also included in the experiment [13]. HB-EGF KO mice showed significantly reduced anastomotic bursting pressure and impaired healing scores on the sixth postoperative day. Moreover, higher mortality and greater complication rates postoperatively were reported in HB-EGF KO mice compared to normal controls. Histopathologically, lower collagen levels, severe inflammation in the submucosal space around the anastomosis, and reduced angiogenesis were noted in KO mice. In contrast, HB-EGF TG mice presented increased anastomotic bursting pressure and higher healing scores on the sixth postoperative day, similar mortality, and lower complication rates postoperatively. Histological examination revealed increased angiogenesis and collagen production, minimal signs of inflammation, improved epithelialization, and significant submucosal healing. Per os administration of HB-EGF showed increased anastomotic bursting pressure and significantly lower complication rates. These findings are supportive of the possible cytoprotective role of HB-EGF for intestinal epithelial cells [83].
5. Conclusions
In conclusion, we have identified numerous genes which play crucial roles in anastomotic healing as evidenced by mouse models. IL-10 plays a role in regulating the immune response to prevent excessive inflammation. FHL-2 gene products and HB-EGF improve the structural integrity of the anastomosis, promoting collagen deposition and reducing the bursting pressure. COX-2 gene products are important for signaling VEGF production, contributing to vascularization of the anastomosis.
Author Contributions
Conceptualization, K.P. and G.G.; methodology, G.K. and M.F.; software, G.G.; validation, E.A., V.G. and A.M.; formal analysis, G.G., E.A. and M.F.; investigation, K.P.; resources, G.G.; data curation, M.F., E.A. and K.K.-K.; writing—original draft preparation, G.G., M.F., G.K. and E.A.; writing—review and editing, K.P.; supervision, K.P., K.K.-K., M.P. and I.G.; project administration, K.P. and I.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created. Data taken from previously published studies have been cited in this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gessler, B.; Eriksson, O.; Angenete, E. Diagnosis, treatment, and consequences of anastomotic leakage in colorectal surgery. Int. J. Color. Dis. 2017, 32, 549–556. [Google Scholar] [CrossRef]
- Naragund, A.V.; Prabhu, R.Y.; Hira, P.; Karegar, M.M.; Khuroo, S.F.; Bapat, R.D.; Kantharia, C.; Supe, A.N. Can preoperative computed tomography scan predict the occurrence of a pancreatic anastomotic leak: A prospective study with clinical, radiological and pathological co-relation. Int. J. Hepatobiliary Pancreat. Dis. 2016, 6, 48–56. [Google Scholar]
- Espín, E.; Ciga, M.A.; Pera, M.; Ortiz, H.; Lujan, J.; Fraccalvieri, D.; Biondo, S.; Ciga, M.A.; Espí, A.; Codina, A.; et al. Oncological outcome following anastomotic leak in rectal surgery. J. Br. Surg. 2015, 102, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Flor-Lorente, B.; Noguera-Aguilar, J.F.; Delgado-Rivilla, S.; García-González, J.M.; Rodriguez-Martín, M.; Salinas-Ortega, L.; Casado, M.Á.; Álvarez, M. The economic impact of anastomotic leak after colorectal cancer surgery. Health Econ. Rev. 2023, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.T.; Maykel, J.A. Defining anastomotic leak and the clinical relevance of leaks. Clin. Colon. Rectal Surg. 2021, 34, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.E.; Friess, H.; Sell, F.; Ricken, L.; Shrikhande, S.; Kulli, C.; Büchler, M.W. Enteral nutrition prolongs delayed gastric emptying in patients after Whipple resection. Am. J. Surg. 2000, 180, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.B.; Barbul, A. Repair of full-thickness bowel injury. Crit. Care Med. 2003, 31, S538–S546. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.K.; Chang, E.Y.; Jobe, B.A. Clinical review: Healing in gastrointestinal anastomoses, part I. Microsurg. Off. J. Int. Microsurg. Soc. Eur. Fed. Soc. Microsurg. 2006, 26, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wahl, W.; Hassdenteufel, A.; Hofer, B.; Junginger, T. Temporary colostomies after procedures on the sigmoid colon and rectum: Are they still justified? Langenbecks Arch. Für Chir. 1997, 382, 149–156. [Google Scholar]
- Seifert, W.F.; Verhofstad, A.A.; Wobbes, T.; Lange, W.; Rijken, P.F.; Van Der Kogel, A.J.; Hendriks, T. Quantitation of angiogenesis in healing anastomoses of the rat colon. Exp. Mol. Pathol. 1997, 64, 31–40. [Google Scholar] [CrossRef]
- National Heart, Lung, and Blood Institute. “Study Quality Assessment Tools,” Quality Assessment of Case Control Studies. Available online: https://www.nhlbi.nih.gov/health-topics/study-quality-assessment-tools (accessed on 31 August 2022).
- Rigby, R.J. A new animal model of postsurgical bowel inflammation and fibrosis: The effect of commensal microflora. Gut 2009, 58, 1104–1112. [Google Scholar] [CrossRef]
- Radulescu, A.; Zhang, H.Y.; Chen, C.L.; Chen, Y.; Zhou, Y.; Yu, X.; Besner, G.E. Heparin-binding EGF-like growth factor promotes intestinal anastomotic healing. J. Surg. Res. 2011, 171, 540–550. [Google Scholar] [CrossRef]
- Reisinger, K.W.; Schellekens, D.H.; Bosmans, J.W.; Boonen, B.; Hulsewé, K.W.; Sastrowijoto, P.; Derikx, J.P.; Grootjans, J.; Poeze, M. Cyclooxygenase-2 Is Essential for Colorectal Anastomotic Healing. Ann. Surg. 2017, 265, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Reischl, S.; Lee, J.H.; Miltschitzky, J.R.E.; Vieregge, V.; Walter, R.L.; Twardy, V.; Kasajima, A.; Friess, H.; Kamaly, N.; Neumann, P.A. AAc2-26-Nanoparticles Induce Resolution of Intestinal Inflammation and Anastomotic Healing via Inhibition of NF-κB Signaling in a Model of Perioperative Colitis. Inflamm. Bowel Dis. 2021, 27, 1379–1393. [Google Scholar] [CrossRef]
- Kirfel, J.; Pantelis, D.; Kabba, M.; Kahl, P.; Röper, A.; Kalff, J.C.; Buettner, R. Impaired intestinal wound healing in Fhl2-deficient mice is due to disturbed collagen metabolism. Exp. Cell Res. 2008, 314, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, Y.; Guo, Z.; Gong, J.; Zhu, W.; Li, N.; Li, J. Triptolide ameliorates ileocolonic anastomosis inflammation in IL-10 deficient mice by mechanism involving suppression of miR-155/SHIP-1 signaling pathway. Mol. Immunol. 2013, 56, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Te Velde, E.A.; Wagenaar, G.T.; Reijerkerk, A.; Roose-Girma, M.; Borel Rinkes, I.H.; Voest, E.E.; Bouma, B.N.; Gebbink, M.F.; Meijers, J.C. Impaired healing of cutaneous wounds and colonic anastomoses in mice lacking thrombin-activatable fibrinolysis inhibitor. J. Thromb. Haemost. 2003, 1, 2087–2096. [Google Scholar] [CrossRef]
- Borowiec, A.M.; Sydora, B.C.; Doyle, J.; Churchill, T.A.; Madsen, K.; Fedorak, R.N. Small bowel fibrosis and systemic inflammatory response after ileocolonic anastomosis in IL-10 null mice. J. Surg. Res. 2012, 178, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D.; Kim, C.S.; Fonkalsrud, E.W.; Zeng, H.; Dindar, H. Effects of chronic corticosteroids and vitamin A on the healing of intestinal anastomoses. Am. J. Surg. 1992, 163, 71–77. [Google Scholar] [CrossRef]
- Caulfield, H.; Hyman, N.H. Anastomotic leak after low anterior resection: A spectrum of clinical entities. JAMA Surg. 2013, 148, 177–182. [Google Scholar] [CrossRef]
- Costedio, M.M.; Hyman, N.H. Outcomes of Colorectal Anastomosis. Semin. Colon. Rectal Surg. 2008, 19, 37–40. [Google Scholar] [CrossRef]
- Branagan, G.; Finnis, D. Prognosis after anastomotic leakage in colorectal surgery. Dis. Colon. Rectum 2005, 48, 1021–1026. [Google Scholar] [CrossRef]
- Bakker, I.S.; Grossmann, I.; Henneman, D.; Havenga, K.; Wiggers, T. Risk factors for anastomotic leakage and leak-related mortality after colonic cancer surgery in a nationwide audit. Br. J. Surg. 2014, 101, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.; Lim, S.; Wan, Y.; Gao, X.; Patkar, A. The burden of gastrointestinal anastomotic leaks: An evaluation of clinical and economic outcomes. J. Gastrointest. Surg. 2014, 18, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Kayama, H.; Jeon, S.G.; Kusu, T.; Isaka, Y.; Rakugi, H.; Yamamoto, M.; Takeda, K. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int. Immunol. 2010, 22, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Chen, L.; Yang, W.; Huang, X.; Ma, C.; Chen, F.; Xiao, Y.; Zhao, Y.; Ma, C.; et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat. Commun. 2018, 9, 3555. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Layunta, E.; Grasa, L.; Pardo, J.; García, S.; Alcalde, A.I.; Mesonero, J.E. Toll-like receptors 2 and 4 modulate intestinal IL-10 differently in ileum and colon. United Eur. Gastroenterol. J. 2018, 6, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Falcon, D.M. The Role of Immune Cells and Cytokines in Intestinal Wound Healing. Int. J. Mol. Sci. 2019, 20, 6097. [Google Scholar] [CrossRef] [PubMed]
- Morhardt, T.L.; Hayashi, A.; Ochi, T.; Quirós, M.; Kitamoto, S.; Nagao-Kitamoto, H.; Kuffa, P.; Atarashi, K.; Honda, K.; Kao, J.Y.; et al. IL-10 produced by macrophages regulates epithelial integrity in the small intestine. Sci. Rep. 2019, 9, 1223. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Hong, S.W.; Han, D.; Yi, J.; Jung, J.; Yang, B.G.; Lee, J.Y.; Lee, M.; Surh, C.D. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 2016, 351, 858–863. [Google Scholar] [CrossRef]
- Girard-Madoux, M.J.; Ober-Blöbaum, J.L.; Costes, L.M.; Kel, J.M.; Lindenbergh-Kortleve, D.J.; Brouwers-Haspels, I.; Heikema, A.P.; Samsom, J.N.; Clausen, B.E. IL-10 control of CD11c+ myeloid cells is essential to maintain immune homeostasis in the small and large intestine. Oncotarget 2016, 7, 32015–32030. [Google Scholar] [CrossRef]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Cardoso, A.; Gil Castro, A.; Martins, A.C.; Carriche, G.M.; Murigneux, V.; Castro, I.; Cumano, A.; Vieira, P.; Saraiva, M. The Dynamics of Interleukin-10-Afforded Protection during Dextran Sulfate Sodium-Induced Colitis. Front. Immunol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef]
- Schreiber, S.; Fedorak, R.N.; Nielsen, O.H.; Wild, G.; Williams, C.N.; Nikolaus, S.; Jacyna, M.; Lashner, B.A.; Gangl, A.; Rutgeerts, P.; et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology 2000, 119, 1461–1472. [Google Scholar]
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.P.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 2006, 4, 754–759. [Google Scholar] [CrossRef]
- Singampalli, K.L.; Balaji, S.; Wang, X.; Parikh, U.M.; Kaul, A.; Gilley, J.; Birla, R.K.; Bollyky, P.L.; Keswani, S.G. The Role of an IL-10/Hyaluronan Axis in Dermal Wound Healing. Front. Cell Dev. Biol. 2020, 8, 636. [Google Scholar] [CrossRef]
- Leung, A.; Crombleholme, T.M.; Keswani, S.G. Fetal wound healing: Implications for minimal scar formation. Curr. Opin. Pediatr. 2012, 24, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Kieran, I.; Knock, A.; Bush, J.; So, K.; Metcalfe, A.; Hobson, R.; Mason, T.; O’Kane, S.; Ferguson, M. Interleukin-10 reduces scar formation in both animal and human cutaneous wounds: Results of two preclinical and phase II randomized control studies. Wound Repair. Regen. 2013, 21, 428–436. [Google Scholar] [CrossRef]
- Yu, C.; Shan, T.; Feng, A.; Li, Y.; Zhu, W.; Xie, Y.; Li, N.; Li, J. Triptolide ameliorates Crohn’s colitis is associated with inhibition of TLRs/NF-κB signaling pathway. Fitoterapia 2011, 82, 709–715. [Google Scholar] [CrossRef]
- Deng, S.; Wang, A.; Chen, X.; Du, Q.; Wu, Y.; Chen, G.; Guo, W.; Li, Y. HBD Inhibits the Development of Colitis-Associated Cancer in Mice via the IL-6R/STAT3 Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 1069. [Google Scholar] [CrossRef]
- Yuan, K.; Li, X.; Lu, Q.; Zhu, Q.; Jiang, H.; Wang, T.; Huang, G.; Xu, A. Application and Mechanisms of Triptolide in the Treatment of Inflammatory Diseases—A Review. Front. Pharmacol. 2019, 10, 1469. [Google Scholar] [CrossRef] [PubMed]
- Futagami, A.; Ishizaki, M.; Fukuda, Y.; Kawana, S.; Yamanaka, N. Wound healing involves induction of cyclooxygenase-2 expression in rat skin. Lab. Investig. 2002, 82, 1503–1513. [Google Scholar] [CrossRef]
- Salgado, F.L.; Artigiani-Neto, R.; Lopes-Filho, G.D.J. Growth factors and cox2 in wound healing: An experimental study with ehrlich tumors. Arq. Bras. Cir. Dig. 2016, 29, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.P.; Shukla, A.; Choudhury, S.; Singh, R.; Anand, M.; Prabhu, S.N. IL1β/TNFα/COX-2/VEGF axis responsible for effective healing potential of C-glucoside xanthone (mangiferin) based ointment in immunocompromised rats. Cytokine 2002, 158, 156012. [Google Scholar] [CrossRef] [PubMed]
- Hatazawa, R.; Tanaka, A.; Tanigami, M.; Amagase, K.; Kato, S.; Ashida, Y.; Takeuchi, K. Cyclooxygenase-2/prostaglandin E2 accelerates the healing of gastric ulcers via EP4 receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G788–G797. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. PPARδ and PGE2 signaling pathways communicate and connect inflammation to colorectal cancer. Inflamm. Cell Signal 2014, 1, 10.14800/ics.338. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riehl, T.E.; Alvarado, D.; Ee, X.; Ciorba, M.A.; Stenson, W.F. Hyaluronic acid promotes Lgr5+ stem cell proliferation and crypt fission through TLR4 and PGE2 transactivation of EGFR. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G63–G73. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Sansom, O.J. The Wae to repair: Prostaglandin E2 (PGE2) triggers intestinal wound repair. EMBO J. 2017, 36, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; VanDussen, K.L.; Malvin, N.P.; Ryu, S.H.; Wang, Y.; Sonnek, N.M.; Lai, C.W.; Stappenbeck, T.S. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 2017, 36, 5–24. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobata, A.; Tanigawa, T.; Nadatani, Y.; Yamagami, H.; Watanabe, K.; Tominaga, K.; Fujiwara, Y.; Takeuchi, K.; Arakawa, T. Activation of the MyD88 signaling pathway inhibits ischemia-reperfusion injury in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G324–G334. [Google Scholar] [CrossRef]
- Okayama, M.; Hayashi, S.; Aoi, Y.; Nishio, H.; Kato, S.; Takeuchi, K. Aggravation by selective COX-1 and COX-2 inhibitors of dextran sulfate sodium (DSS)-induced colon lesions in rats. Dig. Dis. Sci. 2007, 52, 2095–2103. [Google Scholar] [CrossRef]
- Τakeuchi, K.; Tanigami, M.; Amagase, K.; Ochi, A.; Okuda, S.; Hatazawa, R. Endogenous prostaglandin E2 accelerates healing of indomethacin-induced small intestinal lesions through upregulation of vascular endothelial growth factor expression by activation of EP4 receptors. J. Gastroenterol. Hepatol. 2010, 25, S67–S74. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.C.; Lloyd, F.; Lawrance, I.C. Prostaglandin E2 and polyenylphosphatidylcholine protect against intestinal fibrosis and regulate myofibroblast function. Dig. Dis. Sci. 2015, 60, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Arron, M.N.; Lier, E.J.; de Wilt, J.H.; Stommel, M.W.; van Goor, H.; Ten Broek, R.P. Postoperative administration of non-steroidal anti-inflammatory drugs in colorectal cancer surgery does not increase anastomotic leak rate; A systematic review and meta-analysis. Eur. J. Surg. Oncol. 2020, 46, 2167–2217. [Google Scholar] [CrossRef] [PubMed]
- Kastora, S.L.; Osborne, L.L.; Jardine, R.; Kounidas, G.; Carter, B.; Myint, P.K. Non-steroidal anti-inflammatory agents and anastomotic leak rates across colorectal cancer operations and anastomotic sites: A systematic review and meta-analysis of anastomosis specific leak rate and confounding factors. Eur. J. Surg. Oncol. 2021, 47, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Papafili, A.; Hill, M.R.; Brull, D.J.; McAnulty, R.J.; Marshall, R.P.; Humphries, S.E.; Laurent, G.J. Common promoter variant in cyclooxygenase-2 represses gene expression: Evidence of role in acute-phase inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1631–1636. [Google Scholar] [CrossRef]
- Makar, K.W.; Poole, E.M.; Resler, A.J.; Seufert, B.; Curtin, K.; Kleinstein, S.E.; Duggan, D.; Kulmacz, R.J.; Hsu, L.; Whitton, J.; et al. COX-1 (PTGS1) and COX-2 (PTGS2) polymorphisms, NSAID interactions, and risk of colon and rectal cancers in two independent populations. Cancer Causes Control 2013, 24, 2059–2075. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Getting, S.J.; Solito, E.; Murphy, P.M.; Gao, J.L. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am. J. Pathol. 2001, 158, 1969–1973. [Google Scholar] [CrossRef]
- Belvedere, R.; Novizio, N.; Pessolano, E.; Tosco, A.; Eletto, D.; Porta, A.; Campiglia, P.; Perretti, M.; Filippelli, A.; Petrella, A. Heparan sulfate binds the extracellular Annexin A1 and blocks its effects on pancreatic cancer cells. Biochem. Pharmacol. 2020, 182, 114252. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016, 2016, 8239258. [Google Scholar] [CrossRef]
- Babbin, B.A.; Parkos, C.A.; Mandell, K.J.; Winfree, L.M.; Laur, O.; Ivanov, A.I.; Nusrat, A. Annexin 2 regulates intestinal epithelial cell spreading and wound closure through Rho-related signaling. Am. J. Pathol. 2007, 170, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Janmey, P.A.; Stossel, T. P Signaling pathways and cell mechanics involved in wound closure by epithelial cell sheets. Curr. Biol. 2000, 10, 831–838. [Google Scholar] [CrossRef]
- Dalli, J.; Norling, L.V.; Renshaw, D.; Cooper, D.; Leung, K.Y.; Perretti, M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008, 112, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Babbin, B.A.; Lee, W.Y.; Parkos, C.A.; Winfree, L.M.; Akyildiz, A.; Perretti, M.; Nusrat, A. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J. Biol. Chem. 2006, 281, 19588–19599. [Google Scholar] [CrossRef] [PubMed]
- de Paula-Silva, M.; da Rocha, G.H.O.; Broering, M.F.; Queiroz, M.L.; Sandri, S.; Loiola, R.A.; Oliani, S.M.; Vieira, A.; Perretti, M.; Farsky, S.H.P. Formyl Peptide Receptors and Annexin A1: Complementary Mechanisms to Infliximab in Murine Experimental Colitis and Crohn’s Disease. Front. Immunol. 2021, 12, 714138. [Google Scholar] [CrossRef]
- de Paula-Silva, M.; Barrios, B.E.; Macció-Maretto, L.; Sena, A.A.; Farsky, S.H.P.; Correa, S.G.; Oliani, S.M. Role of the protein annexin A1 on the efficacy of anti-TNF treatment in a murine model of acute colitis. Biochem. Pharmacol. 2016, 115, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; He, C.; Lv, X.; Fan, L.; Wang, C.; Remmenga, S.W.; Rodabaugh, K.J.; Yang, L.; Lele, S.M.; Yang, P.; et al. The four and a half LIM domains 2 (FHL2) regulates ovarian granulosa cell tumor progression via controlling AKT1 transcription. Cell Death Dis. 2016, 7, e2297. [Google Scholar] [CrossRef]
- Wixler, V. The role of FHL2 in wound healing and inflammation. FASEB J. 2019, 33, 7799–7809. [Google Scholar] [CrossRef] [PubMed]
- Verset, L.; Feys, L.; Trépant, A.L.; De Wever, O.; Demetter, P. FHL2: A scaffold protein of carcinogenesis, tumour-stroma interactions and treatment response. Histol. Histopathol. 2016, 31, 469–478. [Google Scholar]
- Friedrich, F.W.; Reischmann, S.; Schwalm, A.; Unger, A.; Ramanujam, D.; Münch, J.; Müller, O.J.; Hengstenberg, C.; Galve, E.; Charron, P.; et al. FHL2 expression and variants in hypertrophic cardiomyopathy. Basic. Res. Cardiol. 2014, 109, 451. [Google Scholar] [CrossRef]
- Dao, D.T.; Anez-Bustillos, L.; Adam, R.M.; Puder, M.; Bielenberg, D.R. Heparin-Binding Epidermal Growth Factor-Like Growth Factor as a Critical Mediator of Tissue Repair and Regeneration. Am. J. Pathol. 2018, 188, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.B.; Hinman, C.E.; Luquette, M.H.; Nowicki, P.T.; Besner, G.E. Heparin-binding epidermal growth factor-like growth factor protects rat intestine from ischemia/reperfusion injury. J. Surg. Res. 1999, 87, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, M.J.; Sreejayan, N.; Ren, J.; Du, M. Angiotensin II promotes smooth muscle cell proliferation and migration through release of heparin-binding epidermal growth factor and activation of EGF-receptor pathway. Mol. Cells 2005, 20, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Besner, G.E. Heparin-binding epidermal growth factor-like growth factor promotes enterocyte migration and proliferation in neonatal rats with necrotizing enterocolitis. J. Pediatr. Surg. 2007, 42, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Lara-Marquez, M.; Luquette, M.H.; Glenn, S.; Haque, A.; Besner, G.E. Heparin-binding EGF-like growth factor decreases inducible nitric oxide synthase and nitric oxide production after intestinal ischemia/reperfusion injury. Antioxid. Redox Signal 2001, 3, 919–930. [Google Scholar] [CrossRef]
- El-Assal, O.N.; Radulescu, A.; Besner, G.E. Heparin-binding EGF-like growth factor preserves mesenteric microcirculatory blood flow and protects against intestinal injury in rats subjected to hemorrhagic shock and resuscitation. Surgery 2007, 142, 234–242. [Google Scholar] [CrossRef] [PubMed]
- El-Assal, O.N.; Besner, G.E. HB-EGF enhances restitution after intestinal ischemia/reperfusion via PI3K/Akt and MEK/ERK1/2 activation. Gastroenterology 2005, 129, 609–625. [Google Scholar] [CrossRef]
- Mehta, V.B.; Besner, G.E.; Mehta, V.B.; Besner, G.E. HB-EGF promotes angiogenesis in endothelial cells via PI3-kinase and MAPK signaling pathways. Growth Factors 2007, 25, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, B.; Khailova, L.; Clark, J.A.; Hosseini, D.M.; Arganbright, K.M.; Reynolds, C.A.; Halpern, M.D. Comparison of epidermal growth factor and heparin-binding epidermal growth factor-like growth factor for prevention of experimental necrotizing enterocolitis. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 11–18. [Google Scholar] [CrossRef]
- Banerjee, A.; Schambach, F.; DeJong, C.S.; Hammond, S.M.; Reiner, S.L. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur. J. Immunol. 2010, 40, 225–231. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).