
Dysfunctional High-Density Lipoprotein Cholesterol and Coronary Artery Disease: A Narrative Review

 , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
Inclusion and Exclusion Criteria
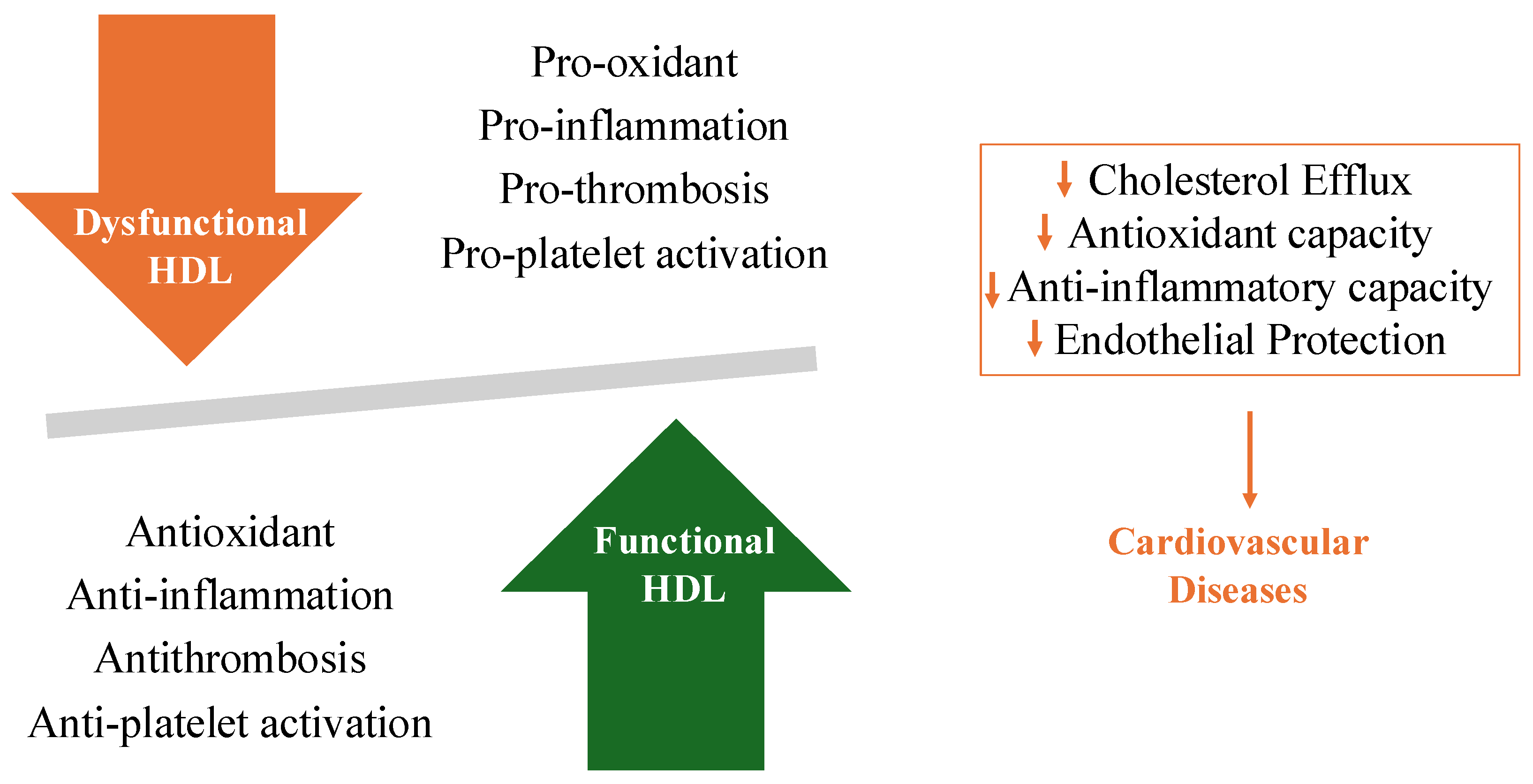
3. Roles of HDL in Protecting against CVD
4. Pathophysiology of Dysfunctional HDL
- It is a structural component of HDL.
- It induces conformational changes necessary for the binding site between pre-β-HDL and membranes.
- It acts as a cofactor for LCAT enzyme activation.
- It is required for the selective uptake of esterified cholesterol into hepatocytes.
5. Role of Dysfunctional HDL on CAD
6. Treatment of Dysfunctional HDL
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saad ALGhasab, N.; Fogacci, F.; Avagimyan, A.; Cicero, A.F.G. Expanding therapeutic options: Overview of novel pharmacotherapies for dyslipidemia. Expert Opin. Pharmacother. 2024. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Atehortua, L.; Davidson, W.S.; Chougnet, C.A. Interactions Between HDL and CD4+ T Cells: A Novel Understanding of HDL Anti-Inflammatory Properties. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1191–1201. [Google Scholar] [CrossRef]
- Liao, J.; Wang, P. Association between paraoxonase 1 -108C/T polymorphism and coronary heart disease: An updated meta-analysis. Front. Cardiovasc. Med. 2024, 11, 1339701. [Google Scholar] [CrossRef]
- Heinecke, J.W. The protein cargo of HDL: Implications for vascular wall biology and therapeutics. J. Clin. Lipidol. 2010, 4, 371–375. [Google Scholar] [CrossRef]
- Primer, K.R.; Psaltis, P.J.; Tan, J.T.M.; Bursill, C.A. The Role of High-Density Lipoproteins in Endothelial Cell Metabolism and Diabetes-Impaired Angiogenesis. Int. J. Mol. Sci. 2020, 21, 3633. [Google Scholar] [CrossRef]
- Sucato, V.; Corrado, E.; Manno, G.; Amata, F.; Testa, G.; Novo, G.; Galassi, A.R. Biomarkers of Coronary Microvascular Dysfunction in Patients with Microvascular Angina: A Narrative Review. Angiology 2022, 73, 395–406. [Google Scholar] [CrossRef]
- Gordon, S.M.; Hofmann, S.; Askew, D.S.; Davidson, W.S. High density lipoprotein: It’s not just about lipid transport anymore. Trends Endocrinol. Metab. 2011, 22, 9–15. [Google Scholar] [CrossRef]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; Derohannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef]
- Sucato, V.; Madaudo, C.; Galassi, A.R. Classification, Diagnosis, and Treatment of Coronary Microvascular Dysfunction. J. Clin. Med. 2022, 11, 4610. [Google Scholar] [CrossRef] [PubMed]
- AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Denimal, D. Antioxidant and Anti-Inflammatory Functions of High-Density Lipoprotein in Type 1 and Type 2 Diabetes. Antioxidants 2024, 13, 57. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Dusting, G.J.; Cutri, B.; Bao, S.; Drummond, G.R.; Rye, K.A.; Barter, P.J. Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 2005, 111, 1543–1550. [Google Scholar] [CrossRef]
- Bale, B.F.; Doneen, A.L.; Leimgruber, P.P.; Vigerust, D.J. The critical issue linking lipids and inflammation: Clinical utility of stopping oxidative stress. Front. Cardiovasc. Med. 2022, 9, 1042729. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Besler, C.; Lüscher, T.F.; Landmesser, U. Molecular mechanisms of vascular effects of High-density lipoprotein: Alterations in cardiovascular disease. EMBO Mol. Med. 2012, 4, 251–268. [Google Scholar] [CrossRef]
- Nofer, J.R.; Levkau, B.; Wolinska, I.; Junker, R.; Fobker, M.; von Eckardstein, A.; Seedorf, U.; Assmann, G. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J. Biol. Chem. 2001, 276, 34480–34485. [Google Scholar] [CrossRef]
- Dib, I.; Khalil, A.; Chouaib, R.; El-Makhour, Y.; Noureddine, H. Apolipoprotein C-III and cardiovascular diseases: When genetics meet molecular pathologies. Mol. Biol. Rep. 2021, 48, 875–886. [Google Scholar] [CrossRef]
- von Eckardstein, A.; Nordestgaard, B.G.; Remaley, A.T.; Catapano, A.L. High-density lipoprotein revisited: Biological functions and clinical relevance. Eur. Heart J. 2023, 44, 1394–1407. [Google Scholar] [CrossRef]
- Sposito, A.C.; de Lima-Junior, J.C.; Moura, F.A.; Barreto, J.; Bonilha, I.; Santana, M.; Virginio, V.W.; Sun, L.; Carvalho, L.S.F.; Soares, A.A.S.; et al. Reciprocal Multifaceted Interaction Between HDL (High-Density Lipoprotein) and Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1550–1564. [Google Scholar] [CrossRef]
- Alboaklah, H.K.M.; McNeish, A.J.; Leake, D.S. Low density lipoprotein oxidized under lysosomal conditions decreases arterial vasodilatation. Free Radic. Res. 2024, 1–8. [Google Scholar] [CrossRef]
- Dastmalchi, L.N.; German, C.A.; Taub, P.R. High density lipoprotein: When to rethink too much of a good thing. Am. J. Prev. Cardiol. 2023, 15, 100511. [Google Scholar] [CrossRef]
- Matsuo, M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef]
- Sucato, V.; Di Fazio, L.; Madaudo, C.; Vadalà, G.; D’Agostino, A.; Evola, S.; Novo, G.; Corrado, E.; Galassi, A.R. Role of Lipoprotein Ratios and Remnant Cholesterol in Patients with Myocardial Infarction with Non-Obstructive Coronary Arteries (MINOCA). J. Cardiovasc. Dev. Dis. 2024, 11, 146. [Google Scholar] [CrossRef]
- Xu, L.B.; Zhou, Y.F.; Yao, J.L.; Sun, S.J.; Rui, Q.; Yang, X.J.; Li, X.B. Apolipoprotein A1 polymorphisms and risk of coronary artery disease: A meta-analysis. Arch. Med. Sci. 2017, 13, 813–819. [Google Scholar] [CrossRef]
- Sucato, V.; Coppola, G.; Testa, G.; Amata, F.; Martello, M.; Siddique, R.; Galassi, A.R.; Novo, G.; Corrado, E. Evaluation of remnant cholesterol levels and Monocyte-to-HDL-cholesterol ratio in South Asian patients with acute coronary syndrome. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2144–2150. [Google Scholar] [CrossRef]
- Sucato, V.; Coppola, G.; Manno, G.; Vadalà, G.; Novo, G.; Corrado, E.; Galassi, A.R. Coronary Artery Disease in South Asian Patients: Cardiovascular Risk Factors, Pathogenesis and Treatments. Curr. Probl. Cardiol. 2023, 48, 101228. [Google Scholar] [CrossRef]
- Franczyk, B.; Rysz, J.; Ławiński, J.; Rysz-Górzyńska, M.; Gluba-Brzózka, A. Is a high hdl-cholesterol level always beneficial? Biomedicines 2021, 9, 1083. [Google Scholar] [CrossRef] [PubMed]
- Glomset, J.A.; Nichols, A.V.; Norum, K.R.; King, W.; Forte, T. Plasma lipoproteins in familial lecithin: Cholesterol acyltransferase defi- ciency. Further studies of very low- and low-density lipoprotein abnormalities. J. Clin. Invest. 1973, 52, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Radulović, S.; Gottschalk, B.; Hörl, G.; Zardoya-Laguardia, P.; Schilcher, I.; Hallström, S.; Vujić, N.; Schmidt, K.; Trieb, M.; Graier, W.F.; et al. Endothelial lipase increases eNOS activating capacity of high-density lipoprotein. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158612. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef]
- Lu, J.; Han, G.; Liu, X.; Chen, B.; Peng, K.; Shi, Y.; Zhang, M.; Yang, Y.; Cui, J.; Song, L.; et al. Association of high-density lipoprotein cholesterol with all-cause and cause-specific mortality in a Chinese population of 3.3 million adults: A prospective cohort study. Lancet Reg. Health West Pac. 2023, 42, 100874. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Adorni, M.P.; Ronda, N.; Bernini, F.; Zimetti, F. High Density Lipoprotein Cholesterol Efflux Capacity and Atherosclerosis in Cardiovascular Disease: Pathophysiological Aspects and Pharmacological Perspectives. Cells 2021, 10, 574. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Bacchetti, T.; Ferretti, G.; Carbone, F.; Ministrini, S.; Montecucco, F.; Jamialahmadi, T.; Sahebkar, A. Dysfunctional High-density Lipoprotein: The Role of Myeloperoxidase and Paraoxonase-1. Curr. Med. Chem. 2020, 28, 2842–2850. [Google Scholar] [CrossRef]
- Carnuta, M.G.; Stancu, C.S.; Toma, L.; Sanda, G.M.; Niculescu, L.S.; Deleanu, M.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; et al. Dysfunctional high-density lipoproteins have distinct composition, diminished anti-inflammatory potential and discriminate acute coronary syndrome from stable coronary artery disease patients. Sci. Rep. 2017, 7, 7295. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Tao, H.; Huang, J.; Yancey, P.G.; Yermalitsky, V.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Zagol-Ikapitte, I.; Ye, F.; Amarnath, V.; et al. Scavenging of reactive dicarbonyls with 2-hydroxybenzylamine reduces atherosclerosis in hypercholesterolemic Ldlr-/-mice. Nat. Commun. 2020, 11, 4084. [Google Scholar] [CrossRef]
- Pitchford, L.M.; Driver, P.M.; Fuller, J.C.; Akers, W.S.; Abumrad, N.N.; Amarnath, V.; Milne, G.L.; Chen, S.-C.; Ye, F.; Roberts, L.J., II.; et al. Safety, tolerability, and pharmacokinetics of repeated oral doses of 2-hydroxybenzylamine acetate in healthy volunteers: A double-blind, randomized, placebo-controlled clinical trial. BMC Pharmacol. Toxicol. 2020, 21, 3. [Google Scholar] [CrossRef]
- Davies, S.S. HDL Function and Atherosclerosis: Reactive Dicarbonyls as Promising Targets of Therapy. Circ. Res. 2023, 132, 1521–1545. [Google Scholar]
- Lüscher, T.F.; Landmesser, U.; von Eckardstein, A.; Fogelman, A.M. High-density lipoprotein: Vascular protective effects, dysfunction, and potential as therapeutic target. Circ. Res. 2014, 114, 171–182. [Google Scholar] [CrossRef]
- Baynham, R.; Weaver, S.R.C.; Rendeiro, C.; Veldhuijzen van Zanten, J.J.C.S. Fat intake impairs the recovery of endothelial function following mental stress in young healthy adults. Front. Nutr. 2023, 10, 1275708. [Google Scholar] [CrossRef]
- Morvaridzadeh, M.; Zoubdane, N.; Heshmati, J.; Alami, M.; Berrougui, H.; Khalil, A. High-Density Lipoprotein Metabolism and Function in Cardiovascular Diseases: What about Aging and Diet Effects? Nutrients 2024, 16, 653. [Google Scholar] [CrossRef]
- Adam, S.; Ho, J.H.; Liu, Y.; Siahmansur, T.; Siddals, K.; Iqbal, Z.; Azmi, S.; Senapati, S.; New, J.; Jeziorska, M.; et al. Bariatric Surgery-induced High-density Lipoprotein Functionality Enhancement Is Associated with Reduced Inflammation. J. Clin. Endocrinol. Metab. 2022, 107, 2182–2194. [Google Scholar] [CrossRef]
- Glavinovic, T.; Thanassoulis, G.; de Graaf, J.; Couture, P.; Hegele, R.A.; Sniderman, A.D. Physiological Bases for the Superiority of Apolipoprotein B Over Low-Density Lipoprotein Cholesterol and Non-High-Density Lipoprotein Cholesterol as a Marker of Cardiovascular Risk. J. Am. Heart Assoc. 2022, 11, e025858. [Google Scholar] [CrossRef]
- Karpouzas, G.A.; Papotti, B.; Ormseth, S.R.; Palumbo, M.; Hernandez, E.; Adorni, M.P.; Zimetti, F.; Budoff, M.J.; Ronda, N. Statins influence the relationship between ATP-binding cassette A1 membrane transporter-mediated cholesterol efflux capacity and coronary atherosclerosis in rheumatoid arthritis. J. Transl. Autoimmun. 2023, 7, 100206. [Google Scholar] [CrossRef]
- Ansell, B.J.; Navab, M.; Hama, S.; Kamranpour, N.; Fonarow, G.; Hough, G.; Rahmani, S.; Mottahedeh, R.; Dave, R.; Reddy, S.T.; et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 2003, 108, 2751–2756. [Google Scholar] [CrossRef]
- Pammer, A.; Klobučar, I.; Stadler, J.T.; Meissl, S.; Habisch, H.; Madl, T.; Frank, S.; Degoricija, V.; Marsche, G. Impaired HDL antioxidant and anti-inflammatory functions are linked to increased mortality in acute heart failure patients. Redox Biol. 2024, 76, 103341. [Google Scholar] [CrossRef]
- Khera, A.V.; Patel, P.J.; Reilly, M.P.; Rader, D.J. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J. Am. Coll. Cardiol. 2013, 62, 1909–1910. [Google Scholar] [CrossRef]
- Haynes, R.; Valdes-Marquez, E.; Hopewell, J.C.; Chen, F.; Li, J.; Parish; Landray, M.J.; Armitage, J.; HPS2-THRIVE Collaborative Group. HPS2-THRIVE Steering Committee members. Serious Adverse Effects of Extended-release Niacin/Laropiprant: Results from the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) Trial. Clin. Ther. 2019, 41, 1767–1777. [Google Scholar] [CrossRef]
- Airan-Javia, S.L.; Wolf, R.L.; Wolfe, M.L.; Tadesse, M.; Mohler, E.; Reilly, M.P. Atheroprotective lipoprotein effects of a niacin-simvastatin combination compared to low- and high-dose simvastatin monotherapy. Am. Heart J. 2009, 157, 687.e1–687.e8. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Wu, M.; Gosmanova, A.K.; Becker, J.O.; Wijsman, E.M.; Brunzell, J.D.; Kahn, S.E.; Knopp, R.H.; Lyons, T.J.; Heinecke, J.W. Low clusterin levels in high-density lipoprotein associate with insulin resistance, obesity, and dyslipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2528–2534. [Google Scholar] [CrossRef]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horva, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar]
- Ballantyne, C.M.; Miller, M.; Niesor, E.J.; Burgess, T.; Kallend, D.; Stein, E.A. Effect of dalcetrapib plus pravastatin on lipoprotein metabolism and high-density lipoprotein composition and function in dyslipidemic patients: Results of a phase IIb dose-ranging study. Am. Heart J. 2012, 163, 515–521e3. [Google Scholar] [CrossRef]
- Ray, K.K.; Ditmarsch, M.; Kallend, D.; Niesor, E.J.; Suchankova, G.; Upmanyu, R.; Anzures-Cabrera, J.; Lehnert, V.; Pauly-Evers, M.; Holme, I.; et al. dal-ACUTE Investigators. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: The dal-ACUTE randomized trial. Eur. Heart J. 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Brewer, H.B.; Kastelein, J.J.; Krueger, K.A.; Wang, M.D.; Shao, M.; Hu, B.; McErlean, E.; Nissen, S.E. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: A randomized controlled trial. JAMA 2011, 306, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Shah, S.; Dansky, H.M.; Davidson, M.; Brinton, E.A.; Gotto, A.M., Jr.; Stepanavage, M.; Liu, S.X.; Gibbons, P.; Ashraf, T.B.; et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N. Engl. J. Med. 2010, 363, 2406–2415. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madaudo, C.; Bono, G.; Ortello, A.; Astuti, G.; Mingoia, G.; Galassi, A.R.; Sucato, V. Dysfunctional High-Density Lipoprotein Cholesterol and Coronary Artery Disease: A Narrative Review. J. Pers. Med. 2024, 14, 996. https://doi.org/10.3390/jpm14090996
Madaudo C, Bono G, Ortello A, Astuti G, Mingoia G, Galassi AR, Sucato V. Dysfunctional High-Density Lipoprotein Cholesterol and Coronary Artery Disease: A Narrative Review. Journal of Personalized Medicine. 2024; 14(9):996. https://doi.org/10.3390/jpm14090996
Chicago/Turabian StyleMadaudo, Cristina, Giada Bono, Antonella Ortello, Giuseppe Astuti, Giulia Mingoia, Alfredo Ruggero Galassi, and Vincenzo Sucato. 2024. "Dysfunctional High-Density Lipoprotein Cholesterol and Coronary Artery Disease: A Narrative Review" Journal of Personalized Medicine 14, no. 9: 996. https://doi.org/10.3390/jpm14090996
APA StyleMadaudo, C., Bono, G., Ortello, A., Astuti, G., Mingoia, G., Galassi, A. R., & Sucato, V. (2024). Dysfunctional High-Density Lipoprotein Cholesterol and Coronary Artery Disease: A Narrative Review. Journal of Personalized Medicine, 14(9), 996. https://doi.org/10.3390/jpm14090996