Antisense Therapy in Neurology



Abstract
:1. Introduction
2. Challenges
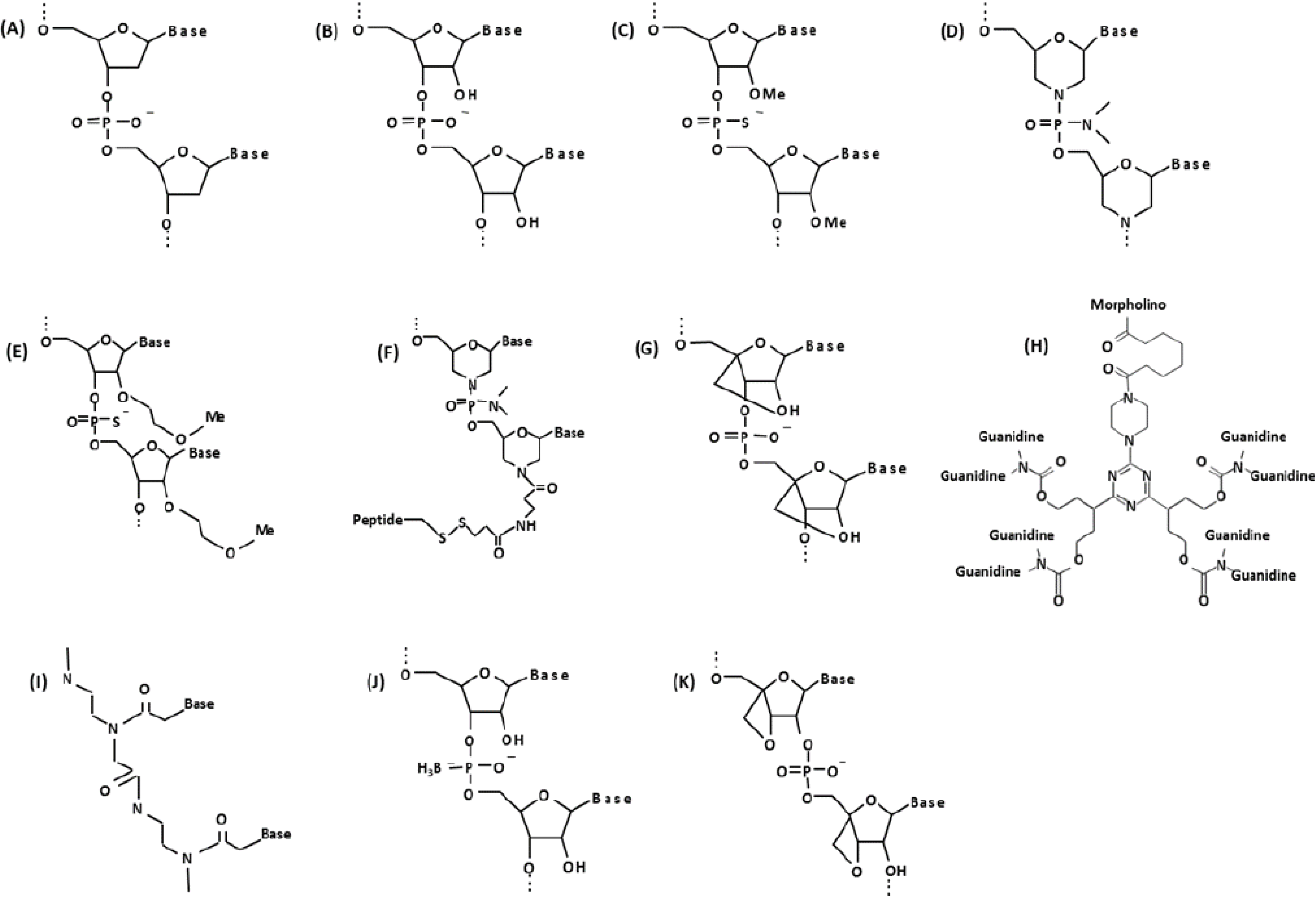
3. Comparative AO Chemistries

4. Antisense Oligo Delivery
5. Antisense Therapy in Neurology: Overview
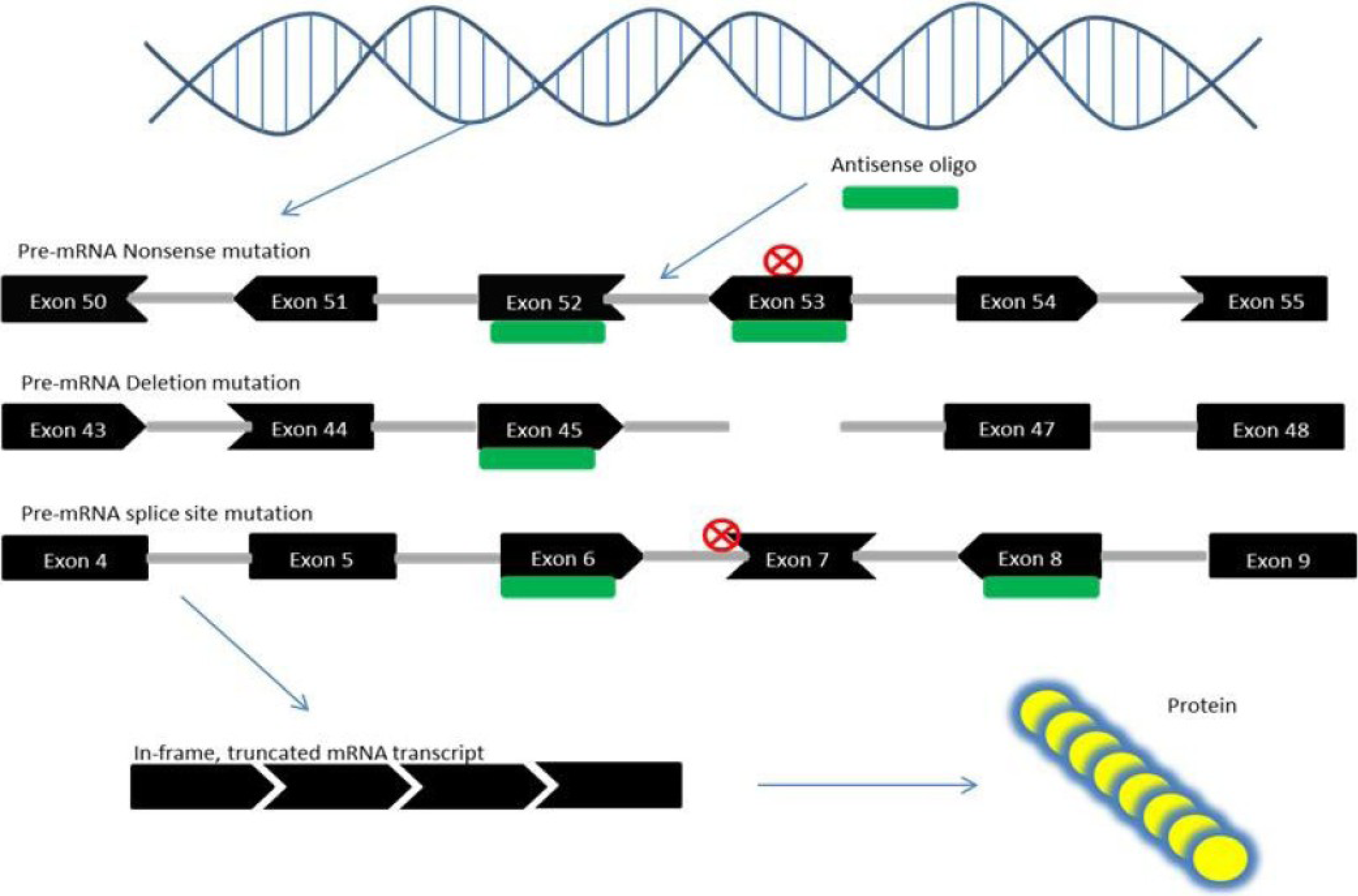
6. Exon Skipping Therapy for DMD


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Drug | Chemistry | Mechanism of action | Target | Clinical Phase | Status | Sponsor | Clinicaltrials.gov ID |
|---|---|---|---|---|---|---|---|---|
| DMD | Eteplirsen (AVI-4658) | PMO | Exon Skipping | Exon 51 | Phase II | Completed | Sarepta Therapeutics | NCT01396239 |
| DMD | Drisapersen (PRO051/GSK2402968) | 2'OMePS | Exon Skipping | Exon 51 | Phase III | Recruiting | Prosensa Therapeutics/ GlaxoSmithKline | NCT01803412 |
| DMD | NS-065/NCNP-01 | PMO | Exon Skipping | Exon 53 | Phase I | Recruiting | Nippon Shinyaku Pharmaceuticals | NA |
| SMA | ISIS-SMNRx | 2'-MOE | Exon Inclusion | Exon 7 | Phase II | Recruiting | Isis Pharmaceuticals | NCT01839656 |
| ALS | ISIS-SOD1Rx/ISIS 333611 | 2'-MOE | Gapmer | Exon 1 | Phase I | Completed | Isis Pharmaceuticals | NCT01041222 |
| DM1 | PRO135 | NA | NA | CUG expansion | Preclinical | In progress | Prosensa Therapeutics | NA |
| HD | PRO289 | NA | NA | CAG expansion | Preclinical | In progress | Prosensa Therapeutics | NA |
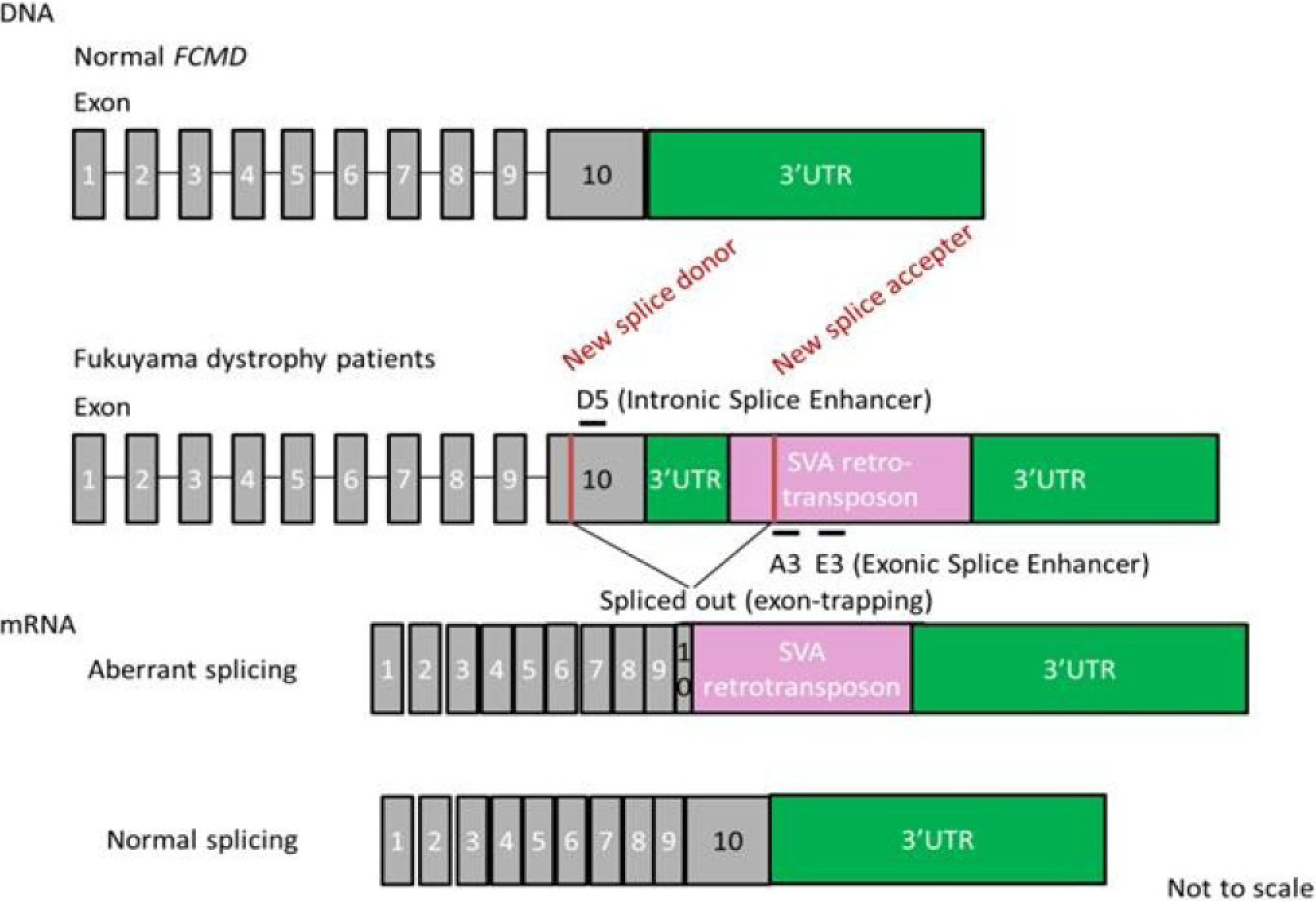
7. Splicing Correction Therapy for FCMD

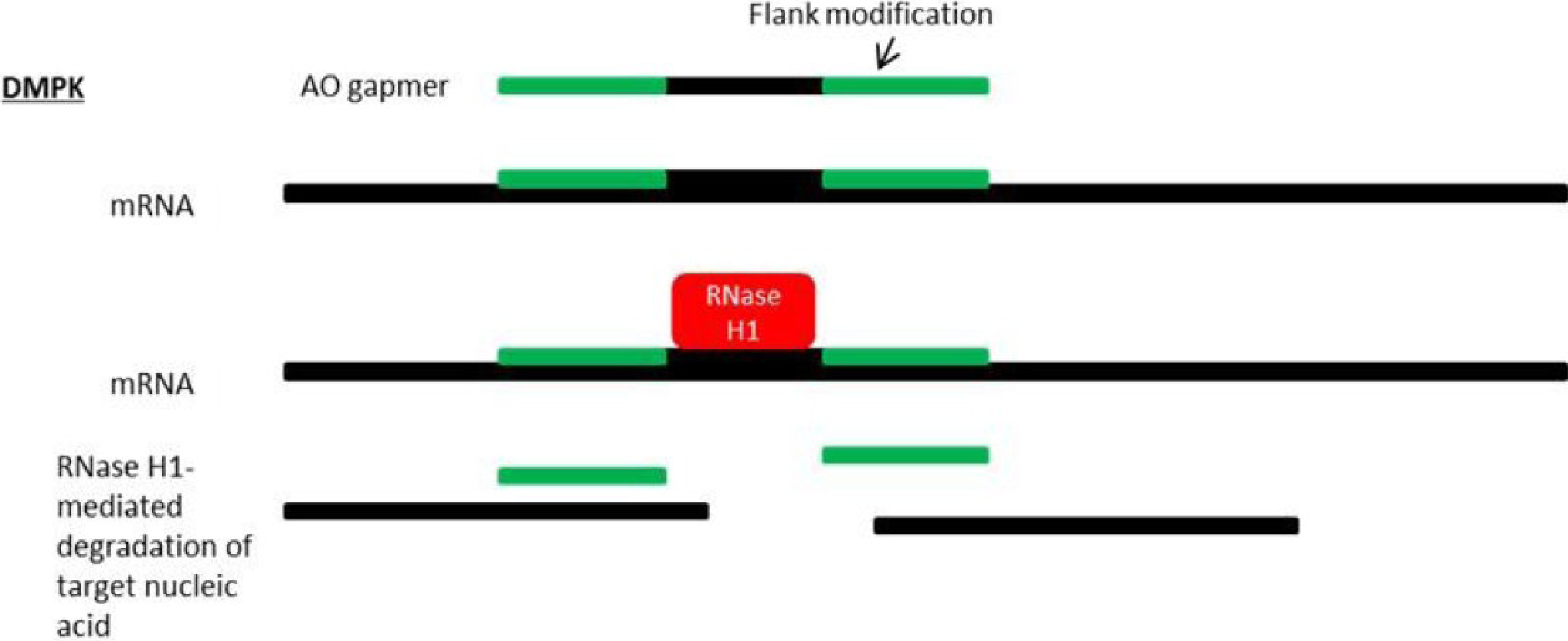
8. Antisense Therapy for DM1

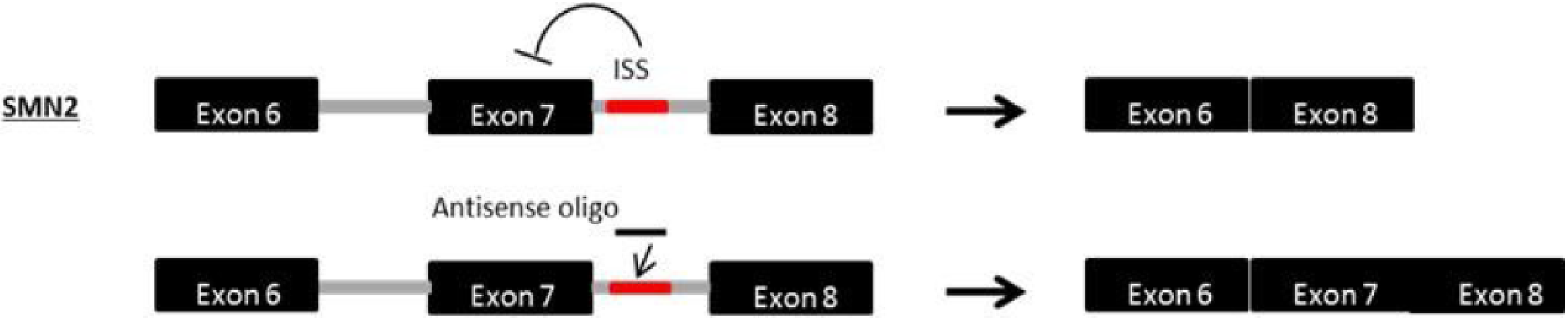
9. Exon Inclusion Therapy for SMA

10. Exon Skipping Therapy for Dysferlinopathy
11. Antisense Therapy for Amyotrophic Lateral Sclerosis (ALS)
12. Antisense Therapy for Huntington’s Disease
13. Conclusions—What Does the Future Hold?
Acknowledgments
Conflict of Interest
References
- Kuzmiak, H.A.; Maquat, L.E. Applying nonsense-mediated mRNA decay research to the clinic: Progress and challenges. Trends Mol. Med. 2006, 12, 306–316. [Google Scholar] [CrossRef]
- Bennett, C.F.; Condon, T.P.; Grimm, S.; Chan, H.; Chiang, M.Y. Inhibition of endothelial cell adhesion molecule expression with antisense oligonucleotides. J. Immunol. 1994, 152, 3530–3540. [Google Scholar]
- Jiang, K. Biotech comes to its “antisenses” after hard-won drug approval. Nat. Med. 2013, 19. [Google Scholar] [CrossRef]
- Bendifallah, N.; Rasmussen, F.W.; Zachar, V.; Ebbesen, P.; Nielsen, P.E.; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug. Chem. 2006, 17, 750–758. [Google Scholar] [CrossRef]
- Miller, P.S.; Braiterman, L.T.; Ts'o, P.O. Effects of a trinucleotide ethyl phosphotriester, Gmp(Et)Gmp(Et)U, on mammalian cells in culture. Biochemistry 1977, 16, 1988–1996. [Google Scholar] [CrossRef]
- Shiraishi, T.; Nielsen, P.E. Improved cellular uptake of antisense peptide nucleic acids by conjugation to a cell-penetrating peptide and a lipid domain. Methods Mol. Biol. 2011, 751, 209–221. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006, 8, 343–375. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef]
- Muntoni, F.; Wood, M.J. Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Bronson, A.; Levin, A.A.; Takeda, S.; Yokota, T.; Baudy, A.R.; Connor, E.M. Restoring dystrophin expression in Duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011, 179, 12–22. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef]
- Broaddus, W.C.; Prabhu, S.S.; Gillies, G.T.; Neal, J.; Conrad, W.S.; Chen, Z.J.; Fillmore, H.; Young, H.F. Distribution and stability of antisense phosphorothioate oligonucleotides in rodent brain following direct intraparenchymal controlled-rate infusion. Neurosurg. Focus 1997, 3, e6. [Google Scholar]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hokfelt, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug delivery to the brain. J. Cereb. Blood Flow Metab. 1997, 17, 713–731. [Google Scholar] [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 2006, 116, 2290–2296. [Google Scholar] [CrossRef]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef]
- Heemskerk, H.A.; de Winter, C.L.; de Kimpe, S.J.; van Kuik-Romeijn, P.; Heuvelmans, N.; Platenburg, G.J.; van Ommen, G.J.; van Deutekom, J.C.; Aartsma-Rus, A. In vivo comparison of 2'-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med. 2009, 11, 257–266. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef]
- Moulton, J.D.; Jiang, S. Gene knockdowns in adult animals: PPMOs and vivo-morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef]
- Sazani, P.; Ness, K.P.; Weller, D.L.; Poage, D.W.; Palyada, K.; Shrewsbury, S.B. Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int. J. Toxicol. 2011, 30, 313–321. [Google Scholar] [CrossRef]
- Altmann, K.H.; Fabbro, D.; Dean, N.M.; Geiger, T.; Monia, B.P.; Muller, M.; Nicklin, P. Second-generation antisense oligonucleotides: Structure-activity relationships and the design of improved signal-transduction inhibitors. Biochem. Soc. Trans. 1996, 24, 630–637. [Google Scholar]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2'-modified oligonucleotides containing 2'-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar]
- Prakash, T.P.; Bhat, B. 2'-Modified oligonucleotides for antisense therapeutics. Curr. Top. Med. Chem. 2007, 7, 641–649. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef]
- Taniguchi-Ikeda, M.; Kobayashi, K.; Kanagawa, M.; Yu, C.C.; Mori, K.; Oda, T.; Kuga, A.; Kurahashi, H.; Akman, H.O.; DiMauro, S.; et al. Pathogenic exon-trapping by SVA retrotransposon and rescue in Fukuyama muscular dystrophy. Nature 2011, 478, 127–131. [Google Scholar] [CrossRef]
- Yokota, T.; Nakamura, A.; Nagata, T.; Saito, T.; Kobayashi, M.; Aoki, Y.; Echigoya, Y.; Partridge, T.; Hoffman, E.P.; Takeda, S. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 2012, 22, 306–315. [Google Scholar]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011, 19, 1295–1303. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.; Betts, C.; Wood, M. CPP-directed oligonucleotide exon skipping in animal models of Duchenne muscular dystrophy. Methods Mol. Biol. 2011, 683, 321–338. [Google Scholar] [CrossRef]
- Yin, H.; Lu, Q.; Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 2008, 16, 38–45. [Google Scholar] [CrossRef]
- Yin, H.; Betts, C.; Saleh, A.F.; Ivanova, G.D.; Lee, H.; Seow, Y.; Kim, D.; Gait, M.J.; Wood, M.J. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Mol. Ther. 2010, 18, 819–827. [Google Scholar] [CrossRef]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Karkare, S.; Bhatnagar, D. Promising nucleic acid analogs and mimics: Characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 2006, 71, 575–586. [Google Scholar] [CrossRef]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef]
- Pfundheller, H.M.; Lomholt, C. Locked nucleic acids: Synthesis and characterization of LNA-T diol. Curr. Protoc. Nucleic Acid Chem. 2002. [Google Scholar] [CrossRef]
- Singh, S.K.; Kumar, R.; Wengel, J. Synthesis of Novel Bicyclo [2.2.1] Ribonucleosides: 2'-Amino- and 2'-Thio-LNA Monomeric Nucleosides. J. Org. Chem. 1998, 63, 6078–6079. [Google Scholar]
- McTigue, P.M.; Peterson, R.J.; Kahn, J.D. Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry 2004, 43, 5388–5405. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, S.K.; Koshkin, A.A.; Rajwanshi, V.K.; Meldgaard, M.; Wengel, J. The first analogues of LNA (locked nucleic acids): Phosphorothioate-LNA and 2'-thio-LNA. Bioorg. Med. Chem. Lett. 1998, 8, 2219–2222. [Google Scholar] [CrossRef]
- Tolstrup, N.; Nielsen, P.S.; Kolberg, J.G.; Frankel, A.M.; Vissing, H.; Kauppinen, S. Oligo Design: Optimal design of LNA (locked nucleic acid) oligonucleotide capture probes for gene expression profiling. Nucleic Acids Res. 2003, 31, 3758–3762. [Google Scholar] [CrossRef]
- Johnson, M.P.; Haupt, L.M.; Griffiths, L.R. Locked nucleic acid (LNA) single nucleotide polymorphism (SNP) genotype analysis and validation using real-time PCR. Nucleic Acids Res. 2004, 32, e55. [Google Scholar] [CrossRef]
- Latorra, D.; Campbell, K.; Wolter, A.; Hurley, J.M. Enhanced allele-specific PCR discrimination in SNP genotyping using 3' locked nucleic acid (LNA) primers. Hum. Mutat. 2003, 22, 79–85. [Google Scholar] [CrossRef]
- Simeonov, A.; Nikiforov, T.T. Single nucleotide polymorphism genotyping using short, fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucleic Acids Res. 2002, 30, e91. [Google Scholar] [CrossRef]
- Petersen, M.; Wengel, J. LNA: A versatile tool for therapeutics and genomics. Trends Biotechnol. 2003, 21, 74–81. [Google Scholar]
- Sood, A.; Spielvogel, B.F.; Shaw, B.R.; Carlton, L.D.; Burnham, B.S.; Hall, E.S.; Hall, I.H. The synthesis and antineoplastic activity of 2'-deoxy-nucleoside-cyanoboranes in murine and human culture cells. Anticancer Res. 1992, 12, 335–343. [Google Scholar]
- Rait, V.K.; Shaw, B.R. Boranophosphates support the RNase H cleavage of polyribonucleotides. Antisense Nucleic Acid Drug Dev. 1999, 9, 53–60. [Google Scholar]
- Rait, V.; Sergueev, D.; Summers, J.; He, K.; Huang, F.; Krzyzanowska, B.; Shaw, B.R. Boranophosphate nucleic acids―A versatile DNA backbone. Nucleos. Nucleot. 1999, 18, 1379–1380. [Google Scholar]
- Li, P.; Shaw, B.R. Synthesis of prodrug candidates: Conjugates of amino acid with nucleoside boranophosphate. Org. Lett. 2002, 4, 2009–2012. [Google Scholar]
- Shaw, B.R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J.L.; Li, P.; Rait, V.; Sergueeva, Z.A.; Sergueev, D. Reading, writing, and modulating genetic information with boranophosphate mimics of nucleotides, DNA, and RNA. Ann. N. Y. Acad. Sci. 2003, 1002, 12–29. [Google Scholar] [CrossRef]
- Shaw, B.R.; Sergueev, D.; He, K.; Porter, K.; Summers, J.; Sergueeva, Z.; Rait, V. Boranophosphate backbone: A mimic of phosphodiesters, phosphorothioates, and methyl phosphonate. Meth. Enzymol. 2000, 313, 226–257. [Google Scholar]
- Opalinska, J.B.; Kalota, A.; Gifford, L.K.; Lu, P.; Jen, K.Y.; Pradeepkumar, P.I.; Barman, J.; Kim, T.K.; Swider, C.R.; Chattopadhyaya, J.; et al. Oxetane modified, conformationally constrained, antisense oligodeoxyribonucleotides function efficiently as gene silencing molecules. Nucleic Acids Res. 2004, 32, 5791–5799. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Gewirtz, A.M. Rationally targeted, conformationally constrained, oxetane-modified oligonucleotides demonstrate efficient gene-silencing activity in a cellular syste. Ann. N. Y. Acad. Sci. 2005, 1058, 39–51. [Google Scholar] [CrossRef]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar]
- Hua, X.; Yu, L.; Huang, X.; Liao, Z.; Xian, Q. Expression and role of fibroblast activation protein-alpha in microinvasive breast carcinoma. Diagn. Pathol. 2011, 6, e111. [Google Scholar]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Sah, D.W.; Aronin, N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Invest. 2011, 121, 500–507. [Google Scholar]
- Heemskerk, H.; de Winter, C.; van Kuik, P.; Heuvelmans, N.; Sabatelli, P.; Rimessi, P.; Braghetta, P.; van Ommen, G.J.; de Kimpe, S.; Ferlini, A.; et al. Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 2010, 18, 1210–1217. [Google Scholar] [CrossRef]
- Kim, Y.; Tewari, M.; Pajeroski, D.J.; Sen, S.; Jason, W.; Sirsi, S.; Lutz, G.; Discher, D.E. Efficient nuclear delivery and nuclear body localization of antisense oligo-nucleotides using degradable polymersomes. In Proceedings of the Engineering in Medicine and Biology Society, 2006. EMBS'06. 28th Annual International Conference of the IEEE, New York, NY, USA, 30 August–3 September 2006; pp. 4350–4353.
- Hoffman, E.P. Skipping toward personalized molecular medicine. N. Engl. J. Med. 2007, 357, 2719–2722. [Google Scholar]
- Yokota, T.; Pistilli, E.; Duddy, W.; Nagaraju, K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2007, 7, 831–842. [Google Scholar]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef]
- Malerba, A.; Boldrin, L.; Dickson, G. Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: Implications for cardiac muscle integrity. Nucleic Acid Ther. 2011, 21, 293–298. [Google Scholar]
- Wheeler, T.M. Myotonic dystrophy: Therapeutic strategies for the future. Neurotherapeutics 2008, 5, 592–600. [Google Scholar]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar]
- Duchenne, G.B. The pathology of paralysis with muscular degeneration (paralysie myosclerotique), or paralysis with apparent hypertrophy. Br. Med. J. 1867, 2, 541–542. [Google Scholar]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar]
- Lu, Q.L.; Yokota, T.; Takeda, S.; Garcia, L.; Muntoni, F.; Partridge, T. The status of exon skipping as a therapeutic approach to Duchenne muscular dystrophy. Mol. Ther. 2011, 19, 9–15. [Google Scholar]
- Aartsma-Rus, A. Antisense-mediated modulation of splicing: Therapeutic implications for Duchenne muscular dystrophy. RNA Biol. 2010, 7, 453–461. [Google Scholar]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing exon skipping therapies for DMD. Acta Myol. 2007, 26, 179–184. [Google Scholar]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar]
- Hoffman, E.P.; Morgan, J.E.; Watkins, S.C.; Partridge, T.A. Somatic reversion/suppression of the mouse mdx phenotype in vivo. J. Neurol. Sci. 1990, 99, 9–25. [Google Scholar] [CrossRef]
- Klein, C.J.; Coovert, D.D.; Bulman, D.E.; Ray, P.N.; Mendell, J.R.; Burghes, A.H. Somatic reversion/suppression in Duchenne muscular dystrophy (DMD): Evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am. J. Hum. Genet. 1992, 50, 950–959. [Google Scholar]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem'yeva, O.V.; Strong, P.; Partridge, T.A. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar]
- Echigoya, Y.; Lee, J.; Rodrigues, M.; Nagata, T.; Tanihata, J.; Nozohourmehrabad, A.; Panesar, D.; Miskew, B.; Aoki, Y.; Yokota, T. Mutation types and aging differently affect revertant fiber expansion in dystrophic Mdx and Mdx52 mice. PLoS One 2013, in press. [Google Scholar]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar]
- Saito, T.; Nakamura, A.; Aoki, Y.; Yokota, T.; Okada, T.; Osawa, M.; Takeda, S. Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS One 2010, 5, e12239. [Google Scholar]
- Aartsma-Rus, A.; de Winter, C.L.; Janson, A.A.; Kaman, W.E.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: Indication for steric hindrance of SR protein binding sites. Oligonucleotides 2005, 15, 284–297. [Google Scholar]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar]
- Bertoni, C.; Lau, C.; Rando, T.A. Restoration of dystrophin expression in mdx muscle cells by chimeraplast-mediated exon skipping. Hum. Mol. Genet. 2003, 12, 1087–1099. [Google Scholar]
- Bremmer-Bout, M.; Aartsma-Rus, A.; de Meijer, E.J.; Kaman, W.E.; Janson, A.A.; Vossen, R.H.; van Ommen, G.J.; den Dunnen, J.T.; van Deutekom, J.C. Targeted exon skipping in transgenic hDMD mice: A model for direct preclinical screening of human-specific antisense oligonucleotides. Mol. Ther. 2004, 10, 232–240. [Google Scholar]
- Fletcher, S.; Honeyman, K.; Fall, A.M.; Harding, P.L.; Johnsen, R.D.; Steinhaus, J.P.; Moulton, H.M.; Iversen, P.L.; Wilton, S.D. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 2007, 15, 1587–1592. [Google Scholar]
- Mann, C.J.; Honeyman, K.; Cheng, A.J.; Ly, T.; Lloyd, F.; Fletcher, S.; Morgan, J.E.; Partridge, T.A.; Wilton, S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 42–47. [Google Scholar]
- McClorey, G.; Fall, A.M.; Moulton, H.M.; Iversen, P.L.; Rasko, J.E.; Ryan, M.; Fletcher, S.; Wilton, S.D. Induced dystrophin exon skipping in human muscle explants. Neuromuscul. Disord. 2006, 16, 583–590. [Google Scholar]
- McClorey, G.; Moulton, H.M.; Iversen, P.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373–1381. [Google Scholar] [CrossRef]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Bremmer-Bout, M.; Janson, A.A.; Ginjaar, I.B.; Baas, F.; den Dunnen, J.T.; van Ommen, G.J. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 2001, 10, 1547–1554. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Takeshima, Y.; Yagi, M.; Wada, H.; Matsuo, M. Intraperitoneal administration of phosphorothioate antisense oligodeoxynucleotide against splicing enhancer sequence induced exon skipping in dystrophin mRNA expressed in mdx skeletal muscle. Brain Dev. 2005, 27, 488–493. [Google Scholar] [CrossRef]
- Yokota, T.; Hoffman, E.; Takeda, S. Antisense oligo-mediated multiple exon skipping in a dog model of Duchenne muscular dystrophy. Methods Mol. Biol. 2011, 709, 299–312. [Google Scholar] [CrossRef]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef]
- Muntoni, F.; Bushby, K.; van Ommen, G. 128th ENMC international workshop on “preclinical optimization and phase I/II clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy” 22–24 October 2004, Naarden, The Netherlands. Neuromuscul. Disord. 2005, 15, 450–457. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Kamoshita, S.; Konishi, Y.; Segawa, M.; Fukuyama, Y. Congenital muscular dystrophy as a disease of the central nervous system. Arch. Neurol. 1976, 33, 513–516. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef]
- Michele, D.E.; Barresi, R.; Kanagawa, M.; Saito, F.; Cohn, R.D.; Satz, J.S.; Dollar, J.; Nishino, I.; Kelley, R.I.; Somer, H.; et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 2002, 418, 417–422. [Google Scholar] [CrossRef]
- Hayashi, Y.K.; Ogawa, M.; Tagawa, K.; Noguchi, S.; Ishihara, T.; Nonaka, I.; Arahata, K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology 2001, 57, 115–121. [Google Scholar] [CrossRef]
- Colombo, R.; Bignamini, A.A.; Carobene, A.; Sasaki, J.; Tachikawa, M.; Kobayashi, K.; Toda, T. Age and origin of the FCMD 3'-untranslated-region retrotransposal insertion mutation causing Fukuyama-type congenital muscular dystrophy in the Japanese population. Hum. genet. 2000, 107, 559–567. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sasaki, J.; Kondo-Iida, E.; Fukuda, Y.; Kinoshita, M.; Sunada, Y.; Nakamura, Y.; Toda, T. Structural organization, complete genomic sequences and mutational analyses of the Fukuyama-type congenital muscular dystrophy gene, fukutin. FEBS Lett. 2001, 489, 192–196. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeo, G.W.; Yeh, R.; Goldstein, P.; Mawson, M.; Sharp, P.A.; Burge, C.B. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004, 32, W187–W190. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef]
- Bhagavati, S.; Leung, B.; Shafiq, S.A.; Ghatpande, A. Myotonic dystrophy: Decreased levels of myotonin protein kinase (Mt-PK) leads to apoptosis in muscle cells. Exp. Neurol. 1997, 146, 277–281. [Google Scholar] [CrossRef]
- Meola, G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve 2000, 23, 1789–1799. [Google Scholar] [CrossRef]
- Meola, G. Myotonic dystrophies. Curr. Opin. Neurol. 2000, 13, 519–525. [Google Scholar] [CrossRef]
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genomics 2010, 11, 77–90. [Google Scholar] [CrossRef]
- Cho, D.H.; Tapscott, S.J. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim. Biophys. Acta 2007, 1772, 195–204. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O'Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3' untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar]
- Hamshere, M.G.; Harley, H.; Harper, P.; Brook, J.D.; Brookfield, J.F. Myotonic dystrophy: The correlation of (CTG) repeat length in leucocytes with age at onset is significant only for patients with small expansions. J. Med. Genet. 1999, 36, 59–61. [Google Scholar]
- Harper, P. Myotonic Dystrophy, 3rd ed.; W B Saunders: London, UK, 2001. [Google Scholar]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef]
- Wang, J.; Pegoraro, E.; Menegazzo, E.; Gennarelli, M.; Hoop, R.C.; Angelini, C.; Hoffman, E.P. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet. 1995, 4, 599–606. [Google Scholar] [CrossRef]
- Magana, J.J.; Cisneros, B. Perspectives on gene therapy in myotonic dystrophy type 1. J. Neurosci. Res. 2011, 89, 275–285. [Google Scholar] [CrossRef]
- Foff, E.P.; Mahadevan, M.S. Therapeutics development in myotonic dystrophy type 1. Muscle Nerve 2011, 44, 160–169. [Google Scholar] [CrossRef]
- Carango, P.; Noble, J.E.; Marks, H.G.; Funanage, V.L. Absence of myotonic dystrophy protein kinase (DMPK) mRNA as a result of a triplet repeat expansion in myotonic dystrophy. Genomics 1993, 18, 340–348. [Google Scholar] [CrossRef]
- Hofmann-Radvanyi, H.; Lavedan, C.; Rabes, J.P.; Savoy, D.; Duros, C.; Johnson, K.; Junien, C. Myotonic dystrophy: Absence of CTG enlarged transcript in congenital forms, and low expression of the normal allele. Hum. Mol. Genet. 1993, 2, 1263–1266. [Google Scholar] [CrossRef]
- Koga, R.; Nakao, Y.; Kurano, Y.; Tsukahara, T.; Nakamura, A.; Ishiura, S.; Nonaka, I.; Arahata, K. Decreased myotonin-protein kinase in the skeletal and cardiac muscles in myotonic dystrophy. Biochem. Biophys. Res. Commun. 1994, 202, 577–585. [Google Scholar] [CrossRef]
- Krahe, R.; Ashizawa, T.; Abbruzzese, C.; Roeder, E.; Carango, P.; Giacanelli, M.; Funanage, V.L.; Siciliano, M.J. Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics 1995, 28, 1–14. [Google Scholar]
- Maeda, M.; Taft, C.S.; Bush, E.W.; Holder, E.; Bailey, W.M.; Neville, H.; Perryman, M.B.; Bies, R.D. Identification, tissue-specific expression, and subcellular localization of the 80- and 71-kDa forms of myotonic dystrophy kinase protein. J. Biol. Chem. 1995, 270, 20246–20249. [Google Scholar]
- Novelli, G.; Gennarelli, M.; Zelano, G.; Pizzuti, A.; Fattorini, C.; Caskey, C.T.; Dallapiccola, B. Failure in detecting mRNA transcripts from the mutated allele in myotonic dystrophy muscle. Biochem. Mol. Biol. Int. 1993, 29, 291–297. [Google Scholar]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Gonzalez-Barriga, A.; Mulders, S.A.; van de Giessen, J.; Hooijer, J.D.; Bijl, S.; van Kessel, I.D.; van Beers, J.; van Deutekom, J.C.; Fransen, J.A.; Wieringa, B.; et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids 2013, 2, e81. [Google Scholar] [CrossRef]
- Lee, J.E.; Bennett, C.F.; Cooper, T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2012, 109, 4221–4226. [Google Scholar]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schoneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- Zellweger, H. The genetic heterogeneity of spinal muscular atrophy (SMA). Birth Defects Orig. Artic. Ser. 1971, 7, 82–89. [Google Scholar]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Khoo, B.; Krainer, A.R. Splicing therapeutics in SMN2 and APOB. Curr. Opin. Mol. Ther. 2009, 11, 108–115. [Google Scholar]
- Markowitz, J.A.; Singh, P.; Darras, B.T. Spinal muscular atrophy: A clinical and research update. Pediatr. Neurol. 2012, 46, 1–12. [Google Scholar] [CrossRef]
- Kolb, S.J.; Kissel, J.T. Spinal muscular atrophy: A timely review. Arch. Neurol. 2011, 68, 979–984. [Google Scholar] [CrossRef]
- Lorson, C.L.; Rindt, H.; Shababi, M. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum. Mol. Genet. 2010, 19, R111–R118. [Google Scholar] [CrossRef]
- van Meerbeke, J.P.; Sumner, C.J. Progress and promise: The current status of spinal muscular atrophy therapeutics. Discov. Med. 2011, 12, 291–305. [Google Scholar]
- Mitrpant, C.; Porensky, P.; Zhou, H.; Price, L.; Muntoni, F.; Fletcher, S.; Wilton, S.D.; Burghes, A.H. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: Towards a treatment for spinal muscular atrophy. PLoS One 2013, 8, e62114. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Aoki, M.; Liu, J.; Richard, I.; Bashir, R.; Britton, S.; Keers, S.M.; Oeltjen, J.; Brown, H.E.; Marchand, S.; Bourg, N.; et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology 2001, 57, 271–278. [Google Scholar]
- Anderson, L.V.; Davison, K.; Moss, J.A.; Young, C.; Cullen, M.J.; Walsh, J.; Johnson, M.A.; Bashir, R.; Britton, S.; Keers, S.; et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 1999, 8, 855–861. [Google Scholar] [CrossRef]
- Argov, Z.; Sadeh, M.; Mazor, K.; Soffer, D.; Kahana, E.; Eisenberg, I.; Mitrani-Rosenbaum, S.; Richard, I.; Beckmann, J.; Keers, S.; et al. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain 2000, 123, 1229–1237. [Google Scholar] [CrossRef]
- Foxton, R.M.; Laval, S.H.; Bushby, K.M. Characterisation of the dysferlin skeletal muscle promoter. Eur. J. Hum. Genet. 2004, 12, 127–131. [Google Scholar] [CrossRef]
- Guglieri, M.; Bushby, K. How to go about diagnosing and managing the limb-girdle muscular dystrophies. Neurol. India 2008, 56, 271–280. [Google Scholar] [CrossRef]
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmuller, H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef]
- Illa, I.; Serrano-Munuera, C.; Gallardo, E.; Lasa, A.; Rojas-Garcia, R.; Palmer, J.; Gallano, P.; Baiget, M.; Matsuda, C.; Brown, R.H. Distal anterior compartment myopathy: A dysferlin mutation causing a new muscular dystrophy phenotype. Ann. Neurol. 2001, 49, 130–134. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar]
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Invest. 2007, 117, 1805–1813. [Google Scholar] [CrossRef]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar]
- Matsuda, C.; Aoki, M.; Hayashi, Y.K.; Ho, M.F.; Arahata, K.; Brown, R.H., Jr. Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology 1999, 53, 1119–1122. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Singh, K.H.; Fokkema, I.F.; Ginjaar, I.B.; van Ommen, G.J.; den Dunnen, J.T.; van der Maarel, S.M. Therapeutic exon skipping for dysferlinopathies? Eur. J. Hum. Genet. 2010, 18, 889–894. [Google Scholar] [CrossRef]
- Wein, N.; Avril, A.; Bartoli, M.; Beley, C.; Chaouch, S.; Laforet, P.; Behin, A.; Butler-Browne, G.; Mouly, V.; Krahn, M.; et al. Efficient bypass of mutations in dysferlin deficient patient cells by antisense-induced exon skipping. Hum. Mutat. 2010, 31, 136–142. [Google Scholar] [CrossRef]
- Sinnreich, M.; Therrien, C.; Karpati, G. Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophy. Neurology 2006, 66, 1114–1116. [Google Scholar] [CrossRef]
- Rothstein, J.D. ALS―Motor neuron disease: Mechanism and development of new therapies. J. Vis. Exp. 2007, e245. [Google Scholar]
- Turner, M.; Al-Chalabi, A. Early symptom progression rate is related to ALS outcome: A prospective population-based study. Neurology 2002, 59, 2012–2013. [Google Scholar] [CrossRef]
- Cleveland, D.W. From Charcot to SOD1: Mechanisms of selective motor neuron death in ALS. Neuron 1999, 24, 515–520. [Google Scholar] [CrossRef]
- Cheah, B.C.; Vucic, S.; Krishnan, A.V.; Boland, R.A.; Kiernan, M.C. Neurophysiological index as a biomarker for ALS progression: Validity of mixed effects models. Amyotroph. Lateral Scler. 2011, 12, 33–38. [Google Scholar] [CrossRef]
- Morariu, M.A. A new classification of amyotrophic lateral sclerosis (ALS) and familial amyotrophic lateral sclerosis (FALS). Dis. Nerv. Syst. 1977, 38, 468–469. [Google Scholar]
- Armani, M.; Pierobon-Bormioli, S.; Mostacciuolo, M.L.; Cacciavillani, M.; Cassol, M.A.; Candeago, R.M.; Angelini, C. Familial ALS: Clinical, genetic and morphological features. Adv. Exp. Med. Biol. 1987, 209, 109–110. [Google Scholar]
- Penco, S.; Schenone, A.; Bordo, D.; Bolognesi, M.; Abbruzzese, M.; Bugiani, O.; Ajmar, F.; Garre, C. A SOD1 gene mutation in a patient with slowly progressing familial ALS. Neurology 1999, 53, 404–406. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar]
- Ratovitski, T.; Corson, L.B.; Strain, J.; Wong, P.; Cleveland, D.W.; Culotta, V.C.; Borchelt, D.R. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum. Mol. Genet. 1999, 8, 1451–1460. [Google Scholar] [CrossRef]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Devos, S.L.; Miller, T.M. Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurotherapeutics 2013, 10, 486–497. [Google Scholar] [CrossRef]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Koppers, M.; van Blitterswijk, M.M.; Vlam, L.; Rowicka, P.A.; van Vught, P.W.; Groen, E.J.; Spliet, W.G.; Engelen-Lee, J.; Schelhaas, H.J.; de Visser, M.; et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 837.e7–837.e13. [Google Scholar]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Mosca, L.; Lunetta, C.; Tarlarini, C.; Avemaria, F.; Maestri, E.; Melazzini, M.; Corbo, M.; Penco, S. Wide phenotypic spectrum of the TARDBP gene: Homozygosity of A382T mutation in a patient presenting with amyotrophic lateral sclerosis, Parkinson’s disease, and frontotemporal lobar degeneration, and in neurologically healthy subject. Neurobiol. Aging 2012, 33, 1846.e1–1846.e4. [Google Scholar]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- van Es, M.A.; Diekstra, F.P.; Veldink, J.H.; Baas, F.; Bourque, P.R.; Schelhaas, H.J.; Strengman, E.; Hennekam, E.A.; Lindhout, D.; Ophoff, R.A.; et al. A case of ALS-FTD in a large FALS pedigree with a K17I ANG mutation. Neurology 2009, 72, 287–288. [Google Scholar] [CrossRef] [Green Version]
- van Langenhove, T.; van der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; van den Broeck, M.; Mattheijssens, M.; Peeters, K.; ve Deyn, P.P.; et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; de Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Byrne, S.; Elamin, M.; Bede, P.; Shatunov, A.; Walsh, C.; Corr, B.; Heverin, M.; Jordan, N.; Kenna, K.; Lynch, C.; et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. 2012, 11, 232–240. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Harms, M.B.; Cady, J.; Zaidman, C.; Cooper, P.; Bali, T.; Allred, P.; Cruchaga, C.; Baughn, M.; Libby, R.T.; Pestronk, A.; et al. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 2234.e13–2234.e19. [Google Scholar]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chio, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Mok, K.Y.; Koutsis, G.; Schottlaender, L.V.; Polke, J.; Panas, M.; Houlden, H. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol. Aging 2012, 33, 1851.e1–1851.e5. [Google Scholar]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013, 21, 102–108. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef]
- Garcia-Redondo, A.; Dols-Icardo, O.; Rojas-Garcia, R.; Esteban-Perez, J.; Cordero-Vazquez, P.; Munoz-Blanco, J.L.; Catalina, I.; Gonzalez-Munoz, M.; Varona, L.; Sarasola, E.; et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum. Mutat. 2013, 34, 79–82. [Google Scholar] [CrossRef]
- Rutherford, N.J.; DeJesus-Hernandez, M.; Baker, M.C.; Kryston, T.B.; Brown, P.E.; Lomen-Hoerth, C.; Boylan, K.; Wszolek, Z.K.; Rademakers, R. C9ORF72 hexanucleotide repeat expansions in patients with ALS from the Coriell Cell Repository. Neurology 2012, 79, 482–483. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Heckman, M.G.; Dejesus-Hernandez, M.; Baker, M.C.; Soto-Ortolaza, A.I.; Rayaprolu, S.; Stewart, H.; Finger, E.; Volkening, K.; Seeley, W.W.; et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 2012, 33, 2950.e5–2950.e7. [Google Scholar]
- Cruts, M.; Gijselinck, I.; van Langenhove, T.; van der Zee, J.; van Broeckhoven, C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013, in press. [Google Scholar]
- Nuytemans, K.; Bademci, G.; Kohli, M.M.; Beecham, G.W.; Wang, L.; Young, J.I.; Nahab, F.; Martin, E.R.; Gilbert, J.R.; Benatar, M.; et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Rademakers, R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012, 11, 297–298. [Google Scholar] [CrossRef]
- Dance, A. Alzheimer Research Forum. In Proceedings of 23rd Annual International Symposium on ALS/MND, Chicago, IL, USA, 5–7 December 2012.
- Donnelly, C.J.; Ostrow, L.W.; Zhang, P.; Vidensky, S.; Hoover, B.N.; Balasubramanian, U.; Li, Y.; Maragakis, N.J.; Tienari, P.; Traynor, B.J.; et al. Development of a C9ORF72 ALS Antisense Therapy and a Therapeutic Biomarker. In Presented at the 2012 Neuroscience Meeting Planner, New Orleans, LA, USA, 17 October 2012.
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Frank, S.; Ondo, W.; Fahn, S.; Hunter, C.; Oakes, D.; Plumb, S.; Marshall, F.; Shoulson, I.; Eberly, S.; Walker, F.; et al. A study of chorea after tetrabenazine withdrawal in patients with Huntington disease. Clin. Neuropharmacol. 2008, 31, 127–133. [Google Scholar] [CrossRef]
- Arnulf, I.; Nielsen, J.; Lohmann, E.; Schiefer, J.; Wild, E.; Jennum, P.; Konofal, E.; Walker, M.; Oudiette, D.; Tabrizi, S.; et al. Rapid eye movement sleep disturbances in Huntington disease. Arch. Neurol. 2008, 65, 482–488. [Google Scholar] [CrossRef]
- Carlock, L.; Walker, P.D.; Shan, Y.; Gutridge, K. Transcription of the Huntington disease gene during the quinolinic acid excitotoxic cascade. Neuroreport 1995, 6, 1121–1124. [Google Scholar] [CrossRef]
- Burns, A.; Folstein, S.; Brandt, J.; Folstein, M. Clinical assessment of irritability, aggression, and apathy in Huntington and Alzheimer disease. J. Nerv. Ment. Dis. 1990, 178, 20–26. [Google Scholar] [CrossRef]
- Marder, K.; Zhao, H.; Myers, R.H.; Cudkowicz, M.; Kayson, E.; Kieburtz, K.; Orme, C.; Paulsen, J.; Penney, J.B., Jr.; Siemers, E.; et al. Rate of functional decline in Huntington’s disease. Neurology 2000, 54, 452–458. [Google Scholar] [CrossRef]
- Reiner, A.; Albin, R.L.; Anderson, K.D.; D'Amato, C.J.; Penney, J.B.; Young, A.B. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5733–5737. [Google Scholar]
- Rosas, H.D.; Hevelone, N.D.; Zaleta, A.K.; Greve, D.N.; Salat, D.H.; Fischl, B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 2005, 65, 745–747. [Google Scholar] [CrossRef]
- Cha, J.H.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef]
- Ross, C.A.; Shoulson, I. Huntington disease: Pathogenesis, biomarkers, and approaches to experimental therapeutics. Parkinsonism Relat. Disord. 2009, 15, S135–S138. [Google Scholar] [CrossRef]
- Liu, J.P.; Zeitlin, S.O. The long and the short of aberrant ciliogenesis in Huntington disease. J. Clin. Invest. 2011, 121, 4237–4241. [Google Scholar] [CrossRef]
- Urbaniak Hunter, K.; Yarbrough, C.; Ciacci, J. Gene- and cell-based approaches for neurodegenerative disease. Adv. Exp. Med. Biol. 2010, 671, 117–130. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Barton, D.E.; Davison, B.C.; Ferguson-Smith, M.A. Analysis of the huntingtin gene reveals a trinucleotide-length polymorphism in the region of the gene that contains two CCG-rich stretches and a correlation between decreased age of onset of Huntington’s disease and CAG repeat number. Hum. Mol. Genet. 1993, 2, 1713–1715. [Google Scholar] [CrossRef]
- Aronin, N.; Chase, K.; Young, C.; Sapp, E.; Schwarz, C.; Matta, N.; Kornreich, R.; Landwehrmeyer, B.; Bird, E.; Beal, M.F.; et al. CAG expansion affects the expression of mutant Huntingtin in the Huntington’s disease brain. Neuron 1995, 15, 1193–1201. [Google Scholar] [CrossRef]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Price, D.L.; Ross, C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar]
- Myers, R.H.; Vonsattel, J.P.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P.; Martin, J.B.; Bird, E.D. Clinical and neuropathologic assessment of severity in Huntington’s disease. Neurology 1988, 38, 341–347. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Gutekunst, C.A.; Persichetti, F.; McNeil, S.M.; Kowall, N.W.; Gusella, J.F.; MacDonald, M.E.; Beal, M.F.; Hersch, S.M. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J. Neurosci. 1997, 17, 3052–3063. [Google Scholar]
- Hoogeveen, A.T.; Willemsen, R.; Meyer, N.; de Rooij, K.E.; Roos, R.A.; van Ommen, G.J.; Galjaard, H. Characterization and localization of the Huntington disease gene product. Hum. Mol. Genet. 1993, 2, 2069–2073. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O'Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- White, J.K.; Auerbach, W.; Duyao, M.P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; MacDonald, M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997, 17, 404–410. [Google Scholar] [CrossRef]
- Rigamonti, D.; Bauer, J.H.; De-Fraja, C.; Conti, L.; Sipione, S.; Sciorati, C.; Clementi, E.; Hackam, A.; Hayden, M.R.; Li, Y.; et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 2000, 20, 3705–3713. [Google Scholar]
- Zhang, Y.; Leavitt, B.R.; van Raamsdonk, J.M.; Dragatsis, I.; Goldowitz, D.; MacDonald, M.E.; Hayden, M.R.; Friedlander, R.M. Huntingtin inhibits caspase-3 activation. EMBO J. 2006, 25, 5896–5906. [Google Scholar] [CrossRef]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelieres, F.P.; de Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Gunawardena, S.; Her, L.S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef]
- Smith, R.; Brundin, P.; Li, J.Y. Synaptic dysfunction in Huntington’s disease: A new perspective. Cell. Mol. Life Sci. 2005, 62, 1901–1912. [Google Scholar] [CrossRef]
- Parker, J.A.; Metzler, M.; Georgiou, J.; Mage, M.; Roder, J.C.; Rose, A.M.; Hayden, M.R.; Neri, C. Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J. Neurosci. 2007, 27, 11056–11064. [Google Scholar] [CrossRef]
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000, 26, 300–306. [Google Scholar] [CrossRef]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Lombardi, M.S.; Jaspers, L.; Spronkmans, C.; Gellera, C.; Taroni, F.; di Maria, E.; Donato, S.D.; Kaemmerer, W.F. A majority of Huntington’s disease patients may be treatable by individualized allele-specific RNA interference. Exp. Neurol. 2009, 217, 312–319. [Google Scholar] [CrossRef]
- Pfister, E.L.; Kennington, L.; Straubhaar, J.; Wagh, S.; Liu, W.; DiFiglia, M.; Landwehrmeyer, B.; Vonsattel, J.P.; Zamore, P.D.; Aronin, N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 2009, 19, 774–778. [Google Scholar] [CrossRef]
- van Bilsen, P.H.; Jaspers, L.; Lombardi, M.S.; Odekerken, J.C.; Burright, E.N.; Kaemmerer, W.F. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum. Gene Ther. 2008, 19, 710–719. [Google Scholar] [CrossRef]
- Hu, J.; Matsui, M.; Corey, D.R. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann. N. Y. Acad. Sci. 2009, 1175, 24–31. [Google Scholar] [CrossRef]
- Hu, J.; Matsui, M.; Gagnon, K.T.; Schwartz, J.C.; Gabillet, S.; Arar, K.; Wu, J.; Bezprozvanny, I.; Corey, D.R. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 2009, 27, 478–484. [Google Scholar] [CrossRef]
- Gagnon, K.T.; Pendergraff, H.M.; Deleavey, G.F.; Swayze, E.E.; Potier, P.; Randolph, J.; Roesch, E.B.; Chattopadhyaya, J.; Damha, M.J.; Bennett, C.F.; et al. Allele-selective inhibition of mutant huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry 2010, 49, 10166–10178. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef]
- Boado, R.J.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Pardridge, W.M. Antisense-mediated down-regulation of the human huntingtin gene. J. Pharmacol. Exp. Ther. 2000, 295, 239–243. [Google Scholar]
- Hu, J.; Dodd, D.W.; Hudson, R.H.; Corey, D.R. Cellular localization and allele-selective inhibition of mutant huntingtin protein by peptide nucleic acid oligomers containing the fluorescent nucleobase [bis-o-(aminoethoxy)phenyl]pyrrolocytosine. Bioorg. Med. Chem. Lett. 2009, 19, 6181–6184. [Google Scholar] [CrossRef]
- Nellemann, C.; Abell, K.; Norremolle, A.; Lokkegaard, T.; Naver, B.; Ropke, C.; Rygaard, J.; Sorensen, S.A.; Hasholt, L. Inhibition of Huntington synthesis by antisense oligodeoxynucleotides. Mol. Cell. Neurosci. 2000, 16, 313–323. [Google Scholar] [CrossRef]
- Fiszer, A.; Olejniczak, M.; Switonski, P.M.; Wroblewska, J.P.; Wisniewska-Kruk, J.; Mykowska, A.; Krzyzosiak, W.J. An evaluation of oligonucleotide-based therapeutic strategies for polyQ diseases. BMC Mol. Biol. 2012, 13, e6. [Google Scholar] [CrossRef]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, J.J.A.; Yokota, T. Antisense Therapy in Neurology. J. Pers. Med. 2013, 3, 144-176. https://doi.org/10.3390/jpm3030144
Lee JJA, Yokota T. Antisense Therapy in Neurology. Journal of Personalized Medicine. 2013; 3(3):144-176. https://doi.org/10.3390/jpm3030144
Chicago/Turabian StyleLee, Joshua J. A., and Toshifumi Yokota. 2013. "Antisense Therapy in Neurology" Journal of Personalized Medicine 3, no. 3: 144-176. https://doi.org/10.3390/jpm3030144
APA StyleLee, J. J. A., & Yokota, T. (2013). Antisense Therapy in Neurology. Journal of Personalized Medicine, 3(3), 144-176. https://doi.org/10.3390/jpm3030144