Transcriptomic Analysis of the Brown Planthopper, Nilaparvata lugens, at Different Stages after Metarhizium anisopliae Challenge



Abstract
:1. Introduction
2. Methods and Materials
2.1. Insect Culture and Fungal Treatment
2.2. Total RNA Isolation, Quantification, and Sequencing
2.3. Data Analysis
2.4. Validation of DEG Libraries Using RT-qPCR
3. Results
3.1. Summary Evaluation of Digital Gene Expression
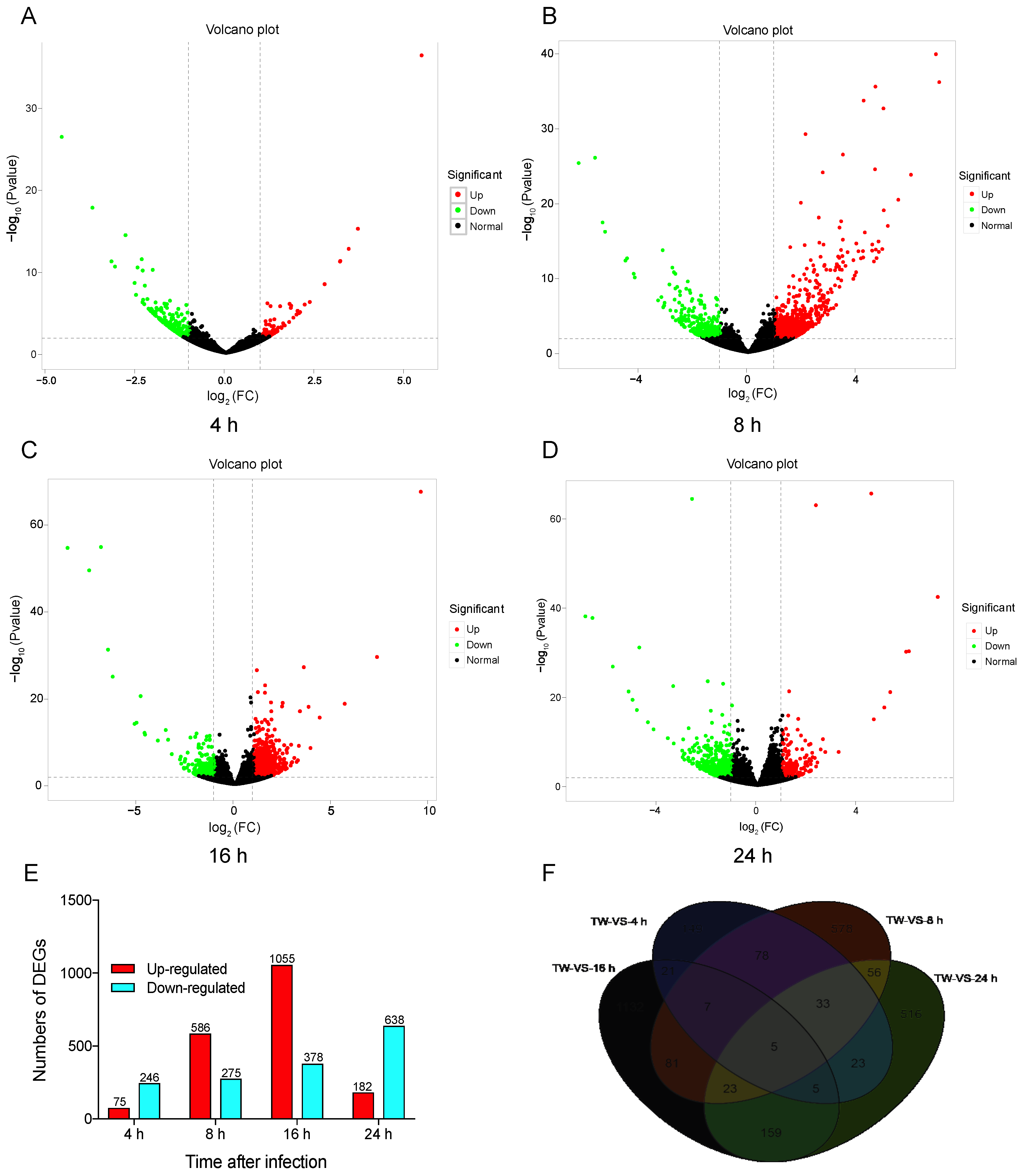
3.2. Transcriptomic Comparison and Analysis of Different Initial Infection Stages
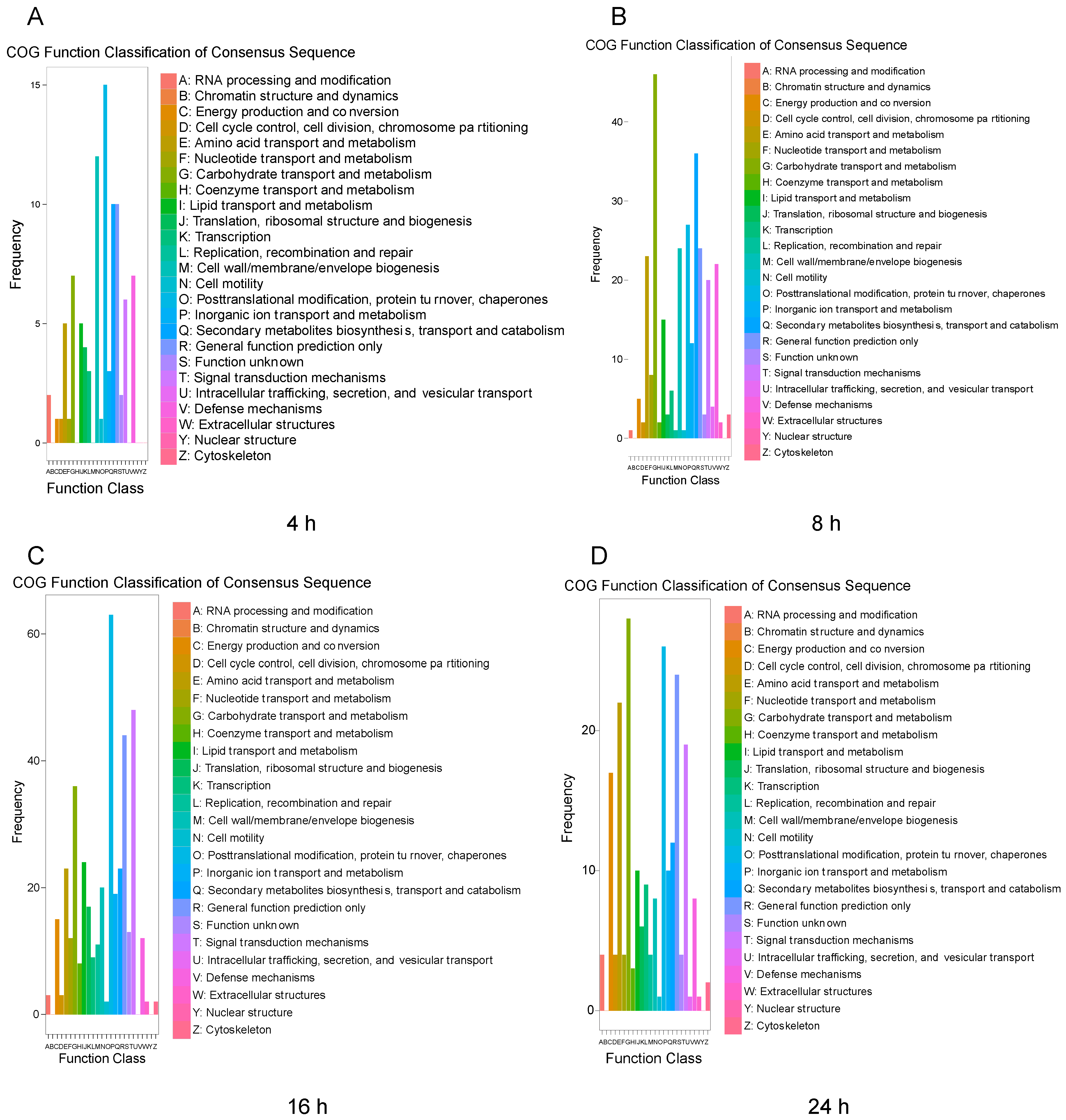
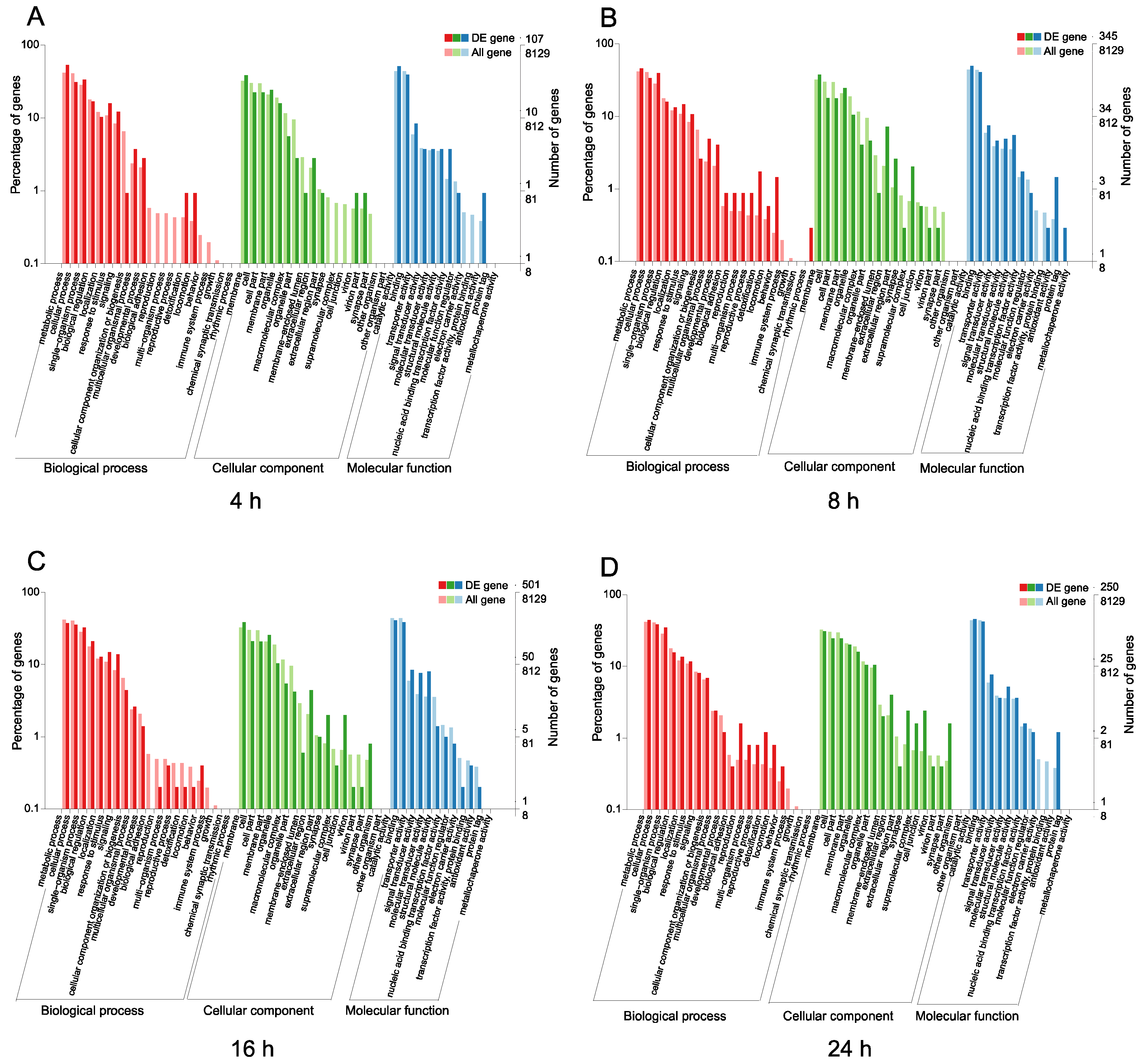
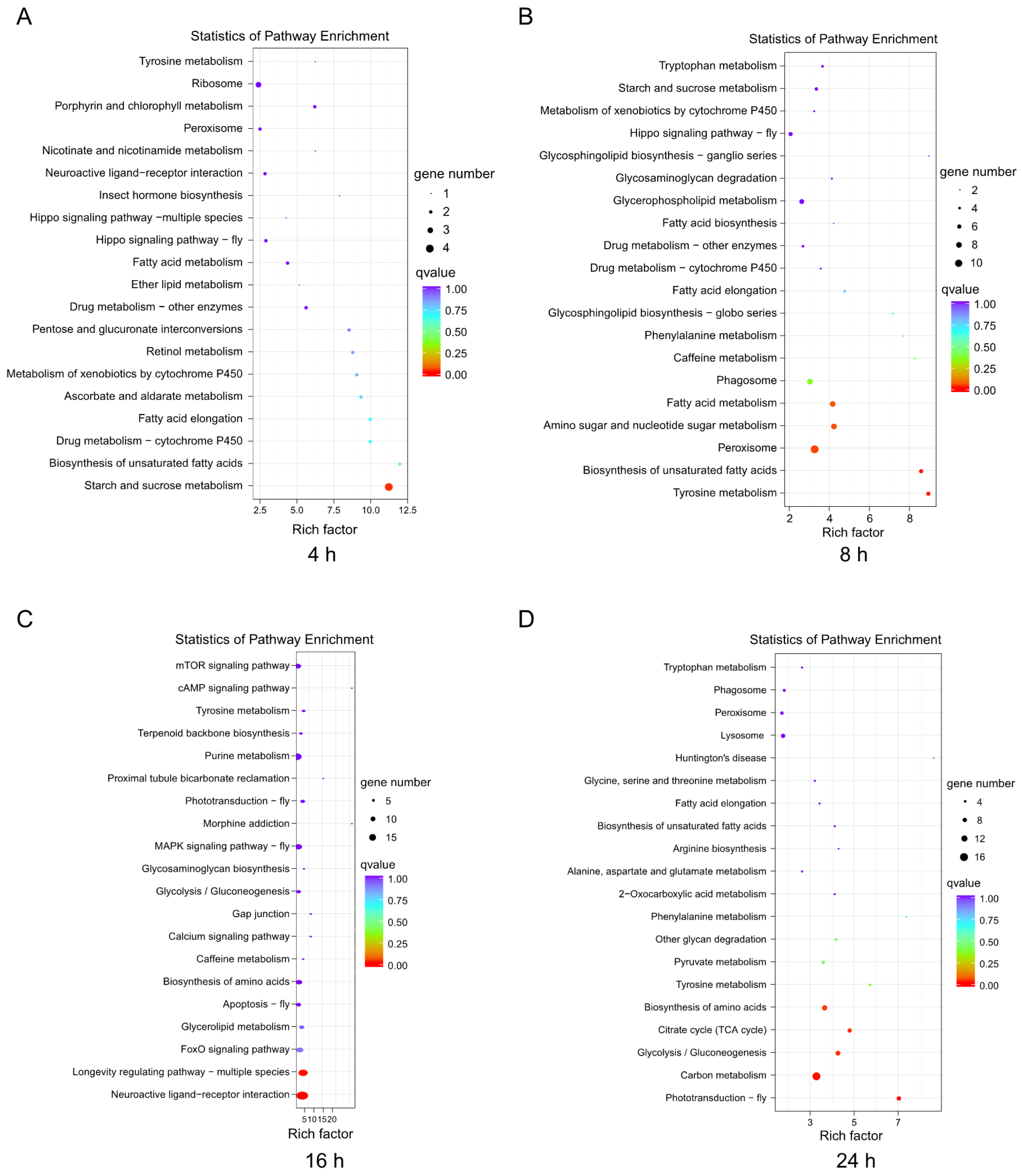
3.3. Functional Classification and Pathway Analysis
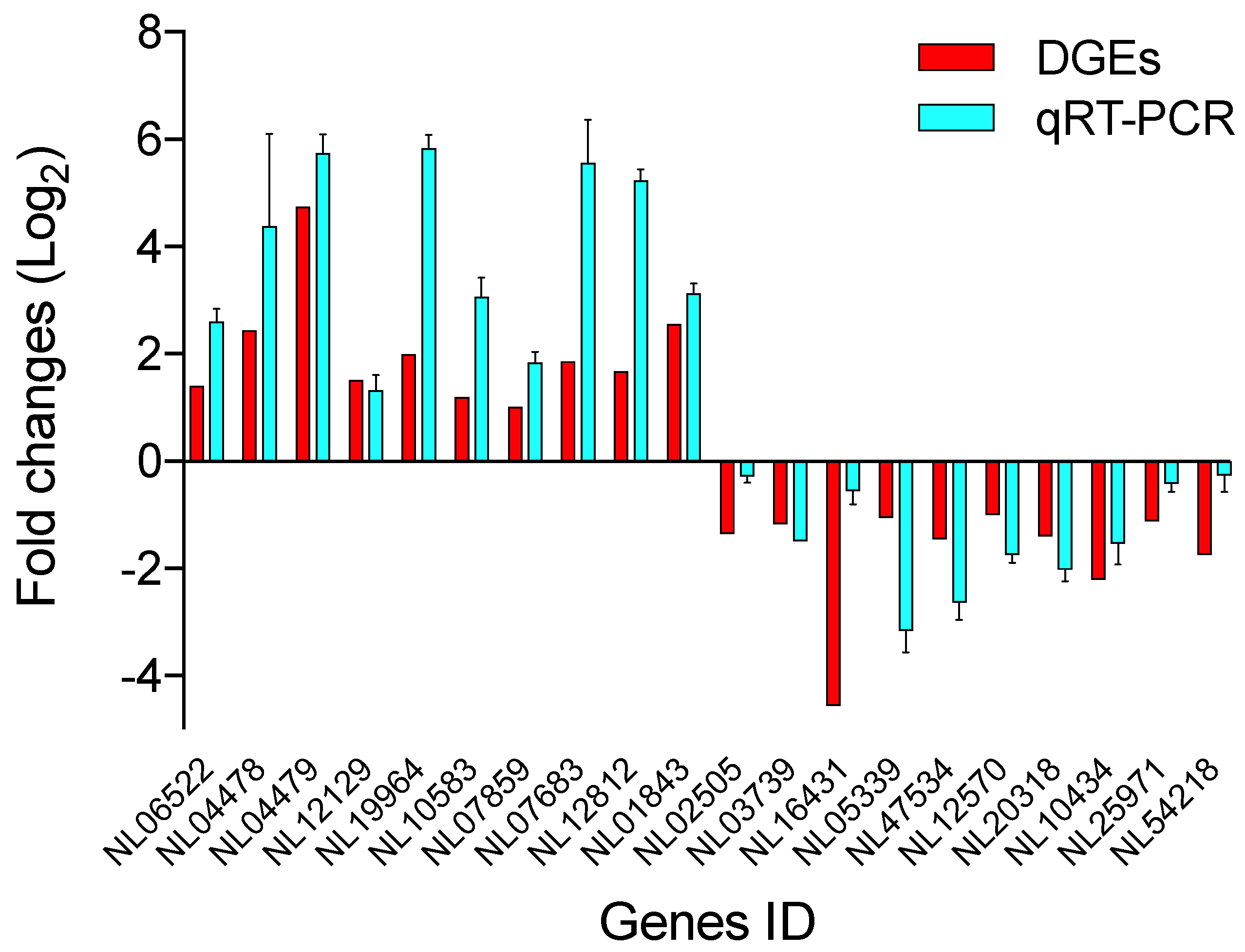
3.4. Validation of DEGs Using RT-qPCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lou, Y.G.; Zhang, G.R.; Zhang, W.Q.; Hu, Y.; Zhang, J. Biological control of rice insect pests in China. Biol. Control 2013, 67, 8–20. [Google Scholar] [CrossRef]
- Cheng, J. Rice planthopper problems and relevant causes in China. In Planthoppers: New Threats to the Sustainability of Intensive Rice Production Systems in Asia; IRRI: Los Banos, Philippines, 2009; pp. 157–178. [Google Scholar]
- Cheng, J. Rice Planthoppers in the Past Half Century in China; Springer: Dordrecht, The Netherlands, 2015; pp. 1–32. [Google Scholar]
- Wang, Y.; Tang, M.; Hao, P.; Yang, Z.; Zhu, L.; He, G. Penetration into rice tissues by brown planthopper and fine structure of the salivary sheaths. Entomol. Exp. Appl. 2010, 129, 295–307. [Google Scholar] [CrossRef]
- Bottrell, D.G.; Schoenly, K.G. Resurrecting the ghost of green revolutions past: The brown planthopper as a recurring threat to high-yielding rice production in tropical Asia. J. Asia-Pac. Entomol. 2012, 15, 122–140. [Google Scholar] [CrossRef]
- Tu, Z.; Ling, B.; Xu, D.; Zhang, M.; Zhou, G. Effects of southern rice black-streaked dwarf virus on the development and fecundity of its vector. Sogatella Furcifera. Virol. J. 2013, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, M.; Sanadamorimura, S. Recent status of insecticide resistance in Asian rice planthoppers. Jpn. Agric. Res. Q. 2012, 44, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, M.; Sanada-Morimura, S.; Otuka, A.; Ohtsu, R.; Sakumoto, S.; Takeuchi, H.; Satoh, M. Insecticide susceptibilities in populations of two rice planthoppers, Nilaparvata lugens and Sogatella furcifera, immigrating into Japan in the period 2005–2012. Pest Manag. Sci. 2014, 70, 615–622. [Google Scholar] [CrossRef]
- Wang, X.Q.; Wang, G.H.; Zhu, Z.R.; Tang, Q.Y.; Hu, Y.; Qiao, F.; Heong, K.L.; Cheng, J.A. Spider (Araneae) predations on white-backed planthopper Sogatella furcifera in subtropical rice ecosystems, China. Pest Manag. Sci. 2017, 73, 1277–1286. [Google Scholar] [CrossRef]
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to green super rice. Nat. Rev. Genet. 2018, 19, 1. [Google Scholar] [CrossRef]
- Guo, J.; Xu, C.; Wu, D.; Zhao, Y.; Qiu, Y.; Wang, X.; Ouyang, Y.; Cai, B.; Liu, X.; Jing, S.; et al. Bph6 encodes an exocyst-localized protein and confers broad resistance to planthoppers in rice. Nat. Genet. 2018, 50, 297–306. [Google Scholar] [CrossRef]
- Tang, W.; Chen, H.; Xu, C.; Li, X.; Lin, Y.; Zhang, Q. Development of insect-resistant transgenic indica rice with a synthetic cry1C* gene. Mol. Breed. 2006, 18, 1–10. [Google Scholar] [CrossRef]
- Hamiduzzaman, M.M.; Sinia, A.; Guzman-Novoa, E.; Goodwin, P.H. Entomopathogenic fungi as potential biocontrol agents of the ecto-parasitic mite, Varroa destructor, and their effect on the immune response of honey bees (Apis mellifera L.). J. Invertebr. Pathol. 2012, 111, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G. Review on safety of the entomopathogenic fungus Metarhizium anisopliae. Biocontrol Sci. Technol. 2007, 17, 879–920. [Google Scholar] [CrossRef]
- Erler, F.; Ates, A.O. Potential of two entomopathogenic fungi, Beauveria bassiana and Metarhizium anisopliae (Coleoptera: Scarabaeidae), as biological control agents against the June beetle. J. Insect Sci. 2015, 15, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.A.; Pell, J.K. Entomopathogenic fungi as biological control agents. Appl. Microbiol. Biotechnol. 2003, 61, 413–423. [Google Scholar] [CrossRef]
- Fite, T.; Tefera, T.; Negeri, M.; Damte, T.; Sori, W. Evaluation of Beauveria bassiana, Metarhizium anisopliae, and Bacillus thuringiensis for the management of Helicoverpa armigera (Hubner) (Lepidoptera: Noctuidae) under laboratory and field conditions. Biocontrol Sci. Technol. 2019. [Google Scholar] [CrossRef]
- Daza, F.F.F.; Roman, G.R.; Rodriguez, M.V.; Vargas, I.A.G.; Heano, H.C.; Cereda, M.P.; Mulet, R.A.C. Spores of Beauveria bassiana and Trichoderma lignorum as a bioinsecticide for the control of Atta cephalotes. Biol. Res. 2019, 52, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rice, S.J.; Baker, D.K.; Leemon, D.M. Development of mycoinsecticide formulations with Beauveria bassiana and Metarhizium anisopliae for the control of lesser mealworm, Alphitobius diaperinus, in chicken broiler houses. Biocontrol 2019, 64, 489–500. [Google Scholar] [CrossRef]
- Jiang, W.; Peng, Y.; Ye, J.; Wen, Y.; Liu, G.; Xie, J. Effects of the entomopathogenic fungus Metarhizium anisopliae on the mortality and immune response of Locusta migratoria. Insects 2019, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Bordalo, M.D.; Gravato, C.; Beleza, S.; Campos, D.; Lopes, I.; Pestana, J.L.T. Lethal and sublethal toxicity assessment of Bacillus thuringiensis var. israelensis and Beauveria bassiana based bioinsecticides to the aquatic insect Chironomus riparius. Sci. Total Environ. 2020, 698, 134155. [Google Scholar]
- Onsongo, S.K.; Gichimu, B.M.; Akutse, K.S.; Dubois, T.; Mohamed, S.A. Performance of three isolates of Metarhizium anisopliae and their virulence against Zeugodacus cucurbitae under different temperature regimes, with global extrapolation of their efficiency. Insects 2019, 10, 270. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Alonso, K.T.; Hernandez-Velazquez, V.M.; Salazar-Schettino, P.M.; Cabrera-Bravo, M.; Toriello, C. Effects of Metarhizium anisopliae on Meccus pallidipennis (Hemiptera: Reduviidae) over different types of wall surfaces. Biocontrol Sci. Technol. 2019, 29, 466–477. [Google Scholar] [CrossRef]
- St Leger, R.; Screen, S. Fungi as Biocontrol Agents: Progress, Problems and Potential; CABI Publishing: Wallingford, UK, 2001. [Google Scholar]
- Farenhorst, M.; Mouatcho, J.C.; Kikankie, C.K.; Brooke, B.D.; Hunt, R.H.; Thomas, M.B.; Koekemoer, L.L.; Knols, B.G.; Coetzee, M. Fungal infection counters insecticide resistance in African malaria mosquitoes. Proc. Natl. Acad. Sci. USA 2009, 106, 17443–17447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farenhorst, M.; Knols, B.G.J.; Thomas, M.B.; Howard, A.F.V.; Takken, W.; Rowland, M.; N’Guessan, R. Synergy in efficacy of fungal entomopathogens and permethrin against west African insecticide-resistant Anopheles gambiae mosquitoes. PLoS ONE 2010, 5, e12081. [Google Scholar] [CrossRef] [PubMed]
- Lovett, B.; Bilgo, E.; Millogo, S.A.; Ouattarra, A.K.; Sare, I.; Gnambani, E.J.; Dabire, R.K.; Diabate, A.; Leger, R.J.S. Transgenic Metarhizium rapidly kills mosquitoes in a malaria-endemic region of Burkina Faso. Science 2019, 364, 894–897. [Google Scholar] [CrossRef]
- Ding, J.L.; Lin, H.Y.; Feng, M.G.; Ying, S.H. Mbp1, a component of the MluI cell cycle box-binding complex, contributes to morphological transition and virulence in the filamentous entomopathogenic fungus Beauveria bassiana. Environ. Microbiol. 2019. [Google Scholar] [CrossRef]
- Peng, G.X.; Xia, Y.X. Integration of an insecticidal scorpion toxin (Bj alpha IT) gene into Metarhizium acridum enhances fungal virulence towards Locusta migratoria manilensis. Pest Manag. Sci. 2015, 71, 58–64. [Google Scholar] [CrossRef]
- Tang, J.F.; Liu, X.Y.; Ding, Y.C.; Jiang, W.J.; Xie, J.Q. Evaluation of Metarhizium anisopliae for rice planthopper control and its synergy with selected insecticides. Crop Prot. 2019, 121, 132–138. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Annu. Rev. Entomol. 2017, 62, 73–90. [Google Scholar] [CrossRef]
- Wang, C.S.; St Leger, R.J. The MAD1 adhesin of Metarhizium anisopliae links adhesion with blastospore production and virulence to insects, and the MAD2 adhesin enables attachment to plants. Eukaryot. Cell 2007, 6, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, J.; Keyhani, N.O.; Zhang, Z.; Li, S.; Xia, Y. Comparative transcriptomic analysis of immune responses of the migratory locust, Locusta migratoria, to challenge by the fungal insect pathogen, Metarhizium Acridum. BMC Genom. 2015, 16, 867. [Google Scholar] [CrossRef] [Green Version]
- Duressa, T.F.; Vanlaer, R.; Huybrechts, R. Locust cellular defense against infections: Sites of pathogen clearance and hemocyte proliferation. Dev. Comp. Immunol. 2015, 48, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Söderhäll, K.; Cerenius, L. Role of the prophenoloxidase-activating system in invertebrate immunity. Curr. Opin. Immunol. 1998, 10, 23–28. [Google Scholar] [CrossRef]
- Arya, B.; Pier Adelchi, R.; Leifer, C.A.; Elena, K.; Alexander, S.; Oleg, C.; Shirakawa, A.K.; Farber, J.M.; Segal, D.M.; Oppenheim, J.J. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 2002, 298, 1025–1029. [Google Scholar]
- Mullen, L.M.; Goldsworthy, G.J. Immune responses of locusts to challenge with the pathogenic fungus Metarhizium or high doses of laminarin. J. Insect Physiol. 2006, 52, 389–398. [Google Scholar] [CrossRef]
- Kounatidis, I.; Ligoxygakis, P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012, 2, 120075. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Xu, X.X.; Shakeel, M.; Li, S.Z.; Wang, S.; Zhou, X.Q.; Yu, J.L.; Xu, X.J.; Yu, X.Q.; Jin, F.L. The entomopathogenic fungi Isaria fumosorosea plays a vital role in suppressing the immune system of Plutella xylostella: RNA-Seq and DGE analysis of immunity-related genes. Front. Microbiol. 2017, 8, 1421. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Suetsugu, Y.; Yamamoto, K.; Noda, H.; Shinoda, T. Transcriptome analysis of neuropeptides and G-protein coupled receptors (GPCRs) for neuropeptides in the brown planthopper Nilaparvata lugens. Peptides 2014, 53, 125–133. [Google Scholar] [CrossRef]
- Guo, S.H.; Yu, L.; Liu, Y.M.; Wang, F.F.; Chen, Y.C.; Wang, Y.; Qiu, B.L.; Sang, W. Digital gene expression profiling in larvae of Tribolium castaneum at different periods post UV-B exposure. Ecotox. Environ. Saf. 2019, 174, 514–523. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Jiang, R.X.; Wu, H.S.; Liu, P.; Xie, J.Q.; He, Y.Y.; Pang, H. Next-generation sequencing-based transcriptome analysis of Cryptolaemus montrouzieri under insecticide stress reveals resistance-relevant genes in ladybirds. Genomics 2012, 100, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.A.; Bao, Y.Y.; Li, B.L.; Cheng, Y.B.; Peng, Z.Y.; Liu, H.; Xu, H.J.; Zhu, Z.R.; Lou, Y.G.; Cheng, J.A.; et al. Transcriptome analysis of the brown planthopper Nilaparvata lugens. PLoS ONE 2010, 5, e14233. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.L.; Li, X.; Wang, F.S.; Han, K.H.; Liu, Z.R.; Fan, L.J.; Hua, H.X.; Cai, W.L.; Yao, Y.J. Candidate detoxification-related genes in brown planthopper, Nilaparvata lugens, in response to beta-asarone based on transcriptomic analysis. Ecotox. Environ. Saf. 2019, 185, 109735. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhou, X.; Zhang, C.X.; Yu, L.L.; Fan, H.W.; Wang, Z.; Xu, H.J.; Xi, Y.; Zhu, Z.R.; Zhou, W.W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.L.; Liu, X.Y.; Zhu, F.X.; Li, J.H.; You, H.; Lu, P. Field evolution of insecticide resistance in the brown planthopper (Nilaparvata lugens Stål) in China. Crop Prot. 2014, 58, 61–66. [Google Scholar] [CrossRef]
- Su, J.; Wang, Z.; Zhang, K.; Tian, X.; Yin, Y.; Zhao, X.; Shen, A.; Gao, C.F. Status of insecticide resistance of the whitebacked planthopper, Sogatella furcifera (Hemiptera: Delphacidae). Fla. Entomol. 2013, 96, 948–956. [Google Scholar] [CrossRef]
- Gupta, S.; Sharma, R.K.; Gupta, R.K.; Sinha, S.R.; Singh, R.; Gajbhiye, V.T. Persistence of new insecticides and their efficacy against insect pests of okra. Bull. Environ. Contam. Tox. 2009, 82, 243–247. [Google Scholar] [CrossRef]
- Peng, G.; Wang, Z.; Yin, Y.; Zeng, D.; Xia, Y. Field trials of Metarhizium anisopliae var. acridum (Ascomycota: Hypocreales) against oriental migratory locusts, Locusta migratoria manilensis (Meyen) in Northern China. Crop Prot. 2008, 27, 1244–1250. [Google Scholar] [CrossRef]
- Zhong, K.; Liu, Z.C.; Wang, J.L.; Liu, X.S. The entomopathogenic fungus Nomuraea rileyi impairs cellular immunity of its host Helicoverpa armigera. Arch. Insect Biochem. Physiol. 2017. [Google Scholar] [CrossRef]
- Lu, H.L.; Leger, R.J. Insect immunity to entomopathogenic fungi. Adv. Genet. 2016, 94, 251–285. [Google Scholar]
- Dan, H. Drosophila immunity: Paths and patterns. Curr. Opin. Immunol. 2003, 15, 12–19. [Google Scholar]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.Y.; Qu, L.Y.; Zhao, D.; Chen, L.B.; Jin, H.Y.; Xu, L.M.; Cheng, J.A.; Zhang, C.X. The genome- and transcriptome-wide analysis of innate immunity in the brown planthopper, Nilaparvata lugens. BMC Genom. 2013, 14, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Lu, C.; Geib, S.M.; Zheng, J.; Wu, S.; Zhang, F.; Liang, G. Characterization of Dendrolimus houi Lajonquiere (Lepidoptera: Lasiocampidae) transcriptome across all life stages. Insects 2019, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clean Reads | Total Reads | GC Content | %≥Q30 |
|---|---|---|---|---|
| T-4h-1 | 21,547,204 | 43,094,408 | 42.41% | 90.35% |
| T-4h-2 | 22,512,422 | 45,024,844 | 39.92% | 90.86% |
| T-4h-3 | 24,148,619 | 48,297,238 | 42.26% | 90.63% |
| T-8h-1 | 38,754,441 | 77,508,882 | 42.62% | 91.54% |
| T-8h-2 | 24,002,510 | 48,005,020 | 41.32% | 90.02% |
| T-8h-3 | 22,122,639 | 44,245,278 | 39.74% | 91.17% |
| T-16h-1 | 23,547,935 | 47,095,870 | 42.30% | 91.52% |
| T-16h-2 | 22,069,447 | 44,138,894 | 40.20% | 91.42% |
| T-16h-3 | 22,239,750 | 44,479,500 | 42.57% | 90.65% |
| T-24h-1 | 25,170,248 | 50,340,496 | 42.54% | 89.66% |
| T-24h-2 | 26,533,253 | 53,066,506 | 41.57% | 90.98% |
| T-24h-3 | 22,340,639 | 44,681,278 | 41.71% | 91.47% |
| W-4h-1 | 24,392,303 | 48,784,606 | 41.81% | 90.56% |
| W-4h-2 | 25,368,822 | 50,737,644 | 41.82% | 90.73% |
| W-4h-3 | 23,584,970 | 47,169,940 | 41.91% | 90.75% |
| W-8h-1 | 21,703,936 | 43,407,872 | 44.07% | 90.44% |
| W-8h-2 | 27,725,344 | 55,450,688 | 44.13% | 89.82% |
| W-8h-3 | 31,397,538 | 62,795,076 | 44.10% | 90.28% |
| W-16h-1 | 21,417,269 | 42,834,538 | 40.71% | 89.96% |
| W-16h-2 | 24,218,605 | 48,437,210 | 39.80% | 91.23% |
| W-16h-3 | 20,903,499 | 41,806,998 | 39.35% | 90.55% |
| W-24h-1 | 22,905,768 | 45,811,536 | 42.11% | 91.31% |
| W-24h-2 | 23,248,816 | 46,497,632 | 40.75% | 90.19% |
| W-24h-3 | 20,628,223 | 41,256,446 | 41.82% | 91.05% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Tang, J.; Xie, J. Transcriptomic Analysis of the Brown Planthopper, Nilaparvata lugens, at Different Stages after Metarhizium anisopliae Challenge. Insects 2020, 11, 139. https://doi.org/10.3390/insects11020139
Peng Y, Tang J, Xie J. Transcriptomic Analysis of the Brown Planthopper, Nilaparvata lugens, at Different Stages after Metarhizium anisopliae Challenge. Insects. 2020; 11(2):139. https://doi.org/10.3390/insects11020139
Chicago/Turabian StylePeng, Yifan, Jifeng Tang, and Jiaqin Xie. 2020. "Transcriptomic Analysis of the Brown Planthopper, Nilaparvata lugens, at Different Stages after Metarhizium anisopliae Challenge" Insects 11, no. 2: 139. https://doi.org/10.3390/insects11020139
APA StylePeng, Y., Tang, J., & Xie, J. (2020). Transcriptomic Analysis of the Brown Planthopper, Nilaparvata lugens, at Different Stages after Metarhizium anisopliae Challenge. Insects, 11(2), 139. https://doi.org/10.3390/insects11020139