1. Introduction
European bark beetle
I. typographus is a secondary pest that affects mainly dead, stressed, and weakened spruce trees (
Picea genus). Under certain environmental conditions, they have massive outbreaks, resulting in their attack of healthy trees, thus becoming a forest pest. It has been proposed that bark beetles’ microbiome plays a key role in the insect’s ecology with the provision of nutrients, inhibition of pathogens, degradation of tree defense compounds, among other probable traits yet to unveil [
1,
2].
The bark beetle
I. typographus is a phloem-feeding species. Its main life cycle occurs inside the inner bark of their host. When environmental conditions and host availability are appropriate, male adults seek for a new host, usually recently tumbled or diseased trees. Once the host is found, pheromones are emitted as a signal call for other individuals to infest the tree. Adult bark beetles perforate the external bark to reach the inner bark, where their life cycle will be developed. Adults excavate galleries, and eggs will be laid. Once larvae are born, they excavate their own galleries, develop, and proceed through all the life stages (pupae, young adult, and adults). Thus, almost all their life cycle develops inside the inner bark, which is mainly composed of cellulose, hemicellulose, and lignin [
1,
3,
4].
Picea trees execute a chemical defense when a beetle attack is detected; this defense is mainly mediated via terpenoid and phenolic compounds. It has been described that beetles must be on an outbreak (mass attack) to surpass a healthy tree’s defense and that the microbiota could assists the beetle to tolerate this tree’s chemical defense by degrading such aromatic compounds [
5,
6]. Both lignocellulosic hydrolysis and aromatic compound degradation via microbiota is theorized as a supplementary energy, nutrient, and/or essential biomolecules source for the beetle [
1,
4]. Some microbes associated with the beetle are able to synthesize a constellation of enzymes—e.g., cellulases, hemicellulases, oxidases, and peroxidases—which hydrolyze those complex molecules into sugars and low molecular weight nutrients, which can be assimilated by the microbes themselves or by the beetle [
1,
4].
Besides that, tree’s phloem, the main feed source for the beetle, has been described as a sugar-rich source but poor in phosphorus, nitrogen, and vitamins, among other relevant nutrients [
7,
8,
9], and it has been suggested that the provision of these scarce substances could be a role of the beetle microbiota [
1]. Phosphorus and nitrogen are essential nutrients for live development. In this scenario, phosphorus could be obtained from biomolecules produced by the tree, which are non-assimilable for the beetle, and nitrogen could be recycled from various sources or fixed from the environment by associated microbes [
1,
2]. Wood is deficient in B-vitamin and beetles cannot synthesize this essential vitamin, which could be synthesized by its microbiome [
8].
On the other hand, some of the organisms that beetles encounter on and in their spruce host are pathogens, including various protists, nematodes, virus, bacteria, and fungi [
10,
11,
12]. It has also been proposed that the microbiota could protect the beetles from these enemies, mainly by producing antimicrobial compounds [
1,
13].
Bark beetles mycobiota have been deeply studied; fungal endosymbionts have been identified, and the role of filamentous fungi and yeast role in beetle ecology has been thoroughly described [
2,
8,
14]. Recently, it has been proposed that the beetle’s bacteriome could also be playing important roles for its host, such as providing nutrients, facilitating the access to the phloem, improving defense to pathogens, or degrading the tree’s aromatic compounds, among others [
1,
2,
3,
4,
5,
6,
13,
15].
It has already been described that the beetle’s microbiome is relatively rich in bacterial diversity, including a wide range of genera [
1,
2,
15,
16], and new bacterial species have been isolated from the interior of bark beetles, e.g., from
Ips beetles:
Erwinia typographi or
Pseudomonas bohemica [
13,
17].
Pseudomonas is a bacterial genus that has been repeatedly described as part of the
Ips microbiome [
4,
13,
15,
16]. The
Pseudomonas genus is known as a cosmopolitan group with a wide metabolic machinery, which includes (but is not limited to) the production of antibiotic compounds, hydrolytic enzymes, siderophore molecules, vitamins, and aromatic compounds degradation enzymes [
1,
4,
18,
19].
The present research is a part of a wider study aiming to decode I. typographus bacteriome and its role in the beetle holobiont. Three Pseudomonas strains were isolated from I. typographus adults (strain CA3A) and larvae (strains C2L11 and C2L12B) and characterized; a preliminary analysis of their 16S rRNA gene sequence made us hypothesize that they could constitute a new species within the genus Pseudomonas. Moreover, their presence in different life stages of the beetle (larvae and adults) suggested that they could be playing an important role in the insect’s ecology. Thus, in this work, we aimed to reveal the taxonomic status of these bacterial strains and to sequence and annotate their genomes in order to mine possible traits related to a role within the bark beetle holobiont. According to a polyphasic taxonomic approach, they seem to constitute a new species within the genus, for which the name of Pseudomonas typographi sp. nov. is proposed and CA3AT is designated as a type strain. Moreover, the genome sequences of these three isolates reveal several metabolic pathways that might be related to the beetle’s ecology. Finally, in vitro tests were done to these strains to evaluate some of the most relevant predicted metabolic capabilities of P. typographi strains, concluding a remarkable capability to aid I. typographus nutrition and resistance to fungal pathogens.
3. Results
3.1. Bacterial Identification
As a part of a broader work aiming to study bacteria associated to bark beetles (unpublished), we isolated strains CA3AT, C2L11, and C2L12B, which, based on the nearly complete (approximately 1400 pb) 16S rRNA gene sequence identification, were identified as Pseudomonas but showed a low similarity with their closest related species P. congelans DSM 14939T (97.4%), P. meliae Ogimi 2T (97.2%), and P. silesiensis A3T (97.2%), indicating that they might conform a new Pseudomonas species.
After genome sequences were obtained, complete 16S rRNA and housekeeping gyrB, rpoB¸ and rpoD sequences were used for further comparison with the closest Pseudomonas type strains.
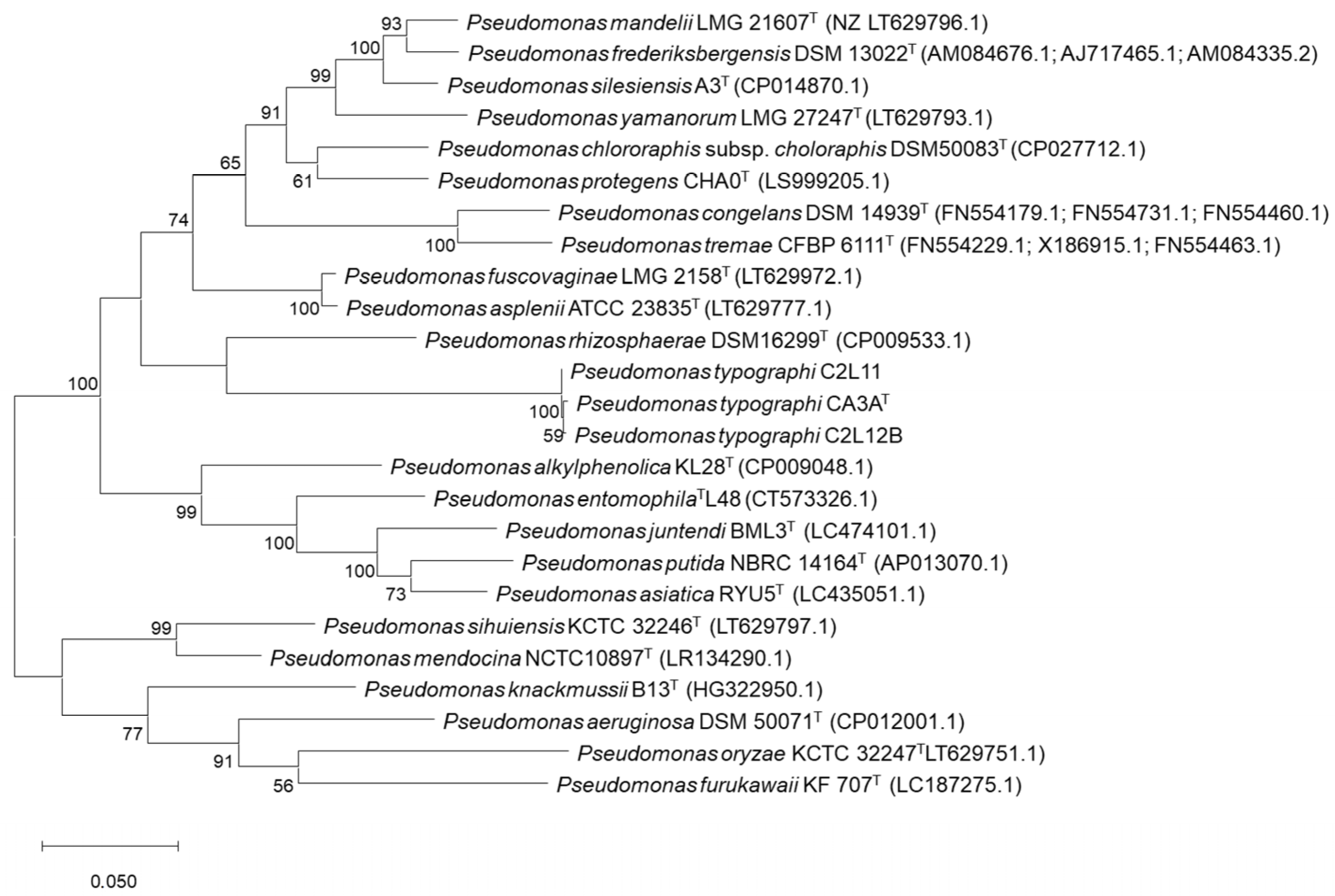
An ML (Maximum Likelihood) phylogenetic tree including all related species within the genus
Pseudomonas that showed an identity over 97% on the 16S rRNA gene sequence was performed.
P. aeruginosa DSM50071
T was included as the type species of the genus and
Permianibacter aggregans HW001
T was included as an outgroup (
Figure 1). This analysis showed that strains CA3A
T, C2L11, and C2L12B cluster together and aside of every closest
Pseudomonas-type strain included in the analysis. As described by Ramírez-Bahena et al. (2014) [
47], analysis based only on the 16S rRNA sequence is very limited to discriminate species within the genus
Pseudomonas at the inter-species level. In consequence, a Multi-Locus Sequence Analysis (MLSA) based on the three housekeeping genes
gyrB,
rpoB, and
rpoD was performed to locate these strains within the genus
Pseudomonas [
47,
48,
49,
50,
51,
52,
53,
54]. ML analysis of the concatenated housekeeping
gyrB,
rpoB, and
rpoD genes sequences (
Figure 2) showed that strains CA3A
T, C2L11, and C2L12B cluster together in an independent branch of any other type strain of
Pseudomonas genus included in the analysis, forming a clade with
P. rhizosphaerae DSM16299
T, which appears in a very distant branch within this clade, suggesting the assignment of the strains of this study to a new species within the genus
Pseudomonas.
To compare the genome sequences of the three strains and the genomes of the closest related species of the genus, ANIb values shared among them were calculated, and a clustered heatmap was performed (
Figure 3). The results showed that the range of similarity of these three strains to the closest
Pseudomonas-type strains is between 75.3% and 77.6%. Most of the related species were
P. entomophila (77.5%),
P. alkylphenolica (77.3%), and
P. rhizosphaerae (77.2%).
An in silico DNA–DNA hybridization (DDH) using the genome-to-genome distance calculator (GGDC) calculator was done to assess (i) if all three strains were, within them, the same
Pseudomonas species and (ii) their relationship to the closest type strains selected of previous phylogenetic analysis. Results showed that the CA3A
T–C2L11 dDDH value is 99.6%, the CA3A
T–C2L12B value is 97.7%, and the C2L11–C2L12B value is 97.7% (identities/HSP length formula), which means that the probability, calculated with a logistic regression, of belonging to the same species is 97.7% or above. Furthermore, all three genomes showed that they are distant enough to be considered a different species to their closest relatives (
Table 2). This analysis also confirmed that
P. entomophila,
P. alkylphenolica, and
P. rhizosphaerae are the closest type strains to
P. typographi CA3A
T, C2L11, and C2L12B (
Table 2).
3.2. Genome Analysis
A genome general description based on RAST analysis is shown in
Table 3. The total genome size of
P. typographi CA3A
T, C2L11, and C2L12B is approximately 5.9–6.1 million pb and the G + C content is 62.0–62.2. The number of contigs obtained after the assembling process of the genome sequences was 229 for CA3A
T, 158 for C2L11, and 174 for C2L12B. The gene calling predicted 5800–6000 coding sequences (CDSs) and 60–64 RNAs sequences for these genomes. Annotated features were classified into 364 subsystems in strains CA3A
T and C2L11B and in 362 for C2L11, understanding a subsystem as a collection of functional roles that are associated to each other in a wider system; approximately 26–27 of the predicted CDSs belong to a subsystem and from these, about 97 are non-hypothetical proteins. Regarding the remaining 74 CDSs, around 41–43 are hypothetical proteins.
According to the KofamKOALA annotations, all three strains isolated in this study were predicted to contain all necessary genes of the metabolic pathway to reduce adenyl sulfate (APS) (EC 2.7.7.4) to 3′-phosphoadenylyl sulfate (PAPS) (EC 2.7.1.25), this to sulfite (EC 1.8.4.8) and finally, to assimilable sulfide (EC 1.8.1.2).
Moreover, the genomes of P. typographi CA3AT, C2L11, and C2L12B have genes annotated as encoding proteins involved in the biosynthesis of thiamine (vit. B1), riboflavin (vit. B2), pyridoxine (vit. B6), nicotinate (vit. B3), biotine (vit. B8), and folate (vit. B9).
Regarding aromatic compounds degradation, all three strains have predicted metabolic pathways related to p-hydroxybenzoic acid (PHBA) degradation. In addition, a gene encoding for the salicylate hydroxylase enzyme (EC 1.14.13.1) was annotated, which catalyzes the conversion of salicylate into catechol.
Secretion systems type II and Sec-SRP were predicted for all three strains. Moreover, for strains C2L11 and C2L12B, gene encoding secretion systems type I and IV were also annotated. These systems were not predicted in the genome sequence of strain CA3AT.
The AntiSMASH program predicted the same five BGCs for all three strains: (i) a bacteriocine cluster that does not resemble any biosynthetic gene cluster (BGC) in the AntiSMASH database but, according to BLASTp, the core gene shows 64.5% similarity to a DUF692 family protein of Shinella kummerowiae, (ii) a Non-Ribosomal Peptide Synthetase-Like (NRPS-Like) cluster, in which 40% of genes show similarity to L-2-amino-4-methoxy-trans-3-butenoic acid (AMB) from P. aeruginosa PAO1, (iii) a NRPS cluster in which 34% of genes show similarity to a crochelin A biosynthetic gene cluster from Azotobacter chroococcum NCIMB 8003, (iv) a NAGGN cluster that does not resemble any known cluster in antiSMASH database but, according to the BLASTp comparison, its three core biosynthetic genes are similar to an N-acetylglutaminylglutamine synthetase (88.4% similarity to Pseudomonas massiliensis), an N-acetylglutaminylglutamine amidotransferase (88.3% similarity to Pseudomonas fluorescens), and an osmoprotectant N-acetylglutaminylglutamine amide (NAGGN) system M42 family peptidase (87.8% similarity to Pseudomonas fluorescens), respectively, and (v) a cluster related to the biosynthesis of a carotenoid molecule, in which 100% of the genes show similarity to that of Enterobacteriaceae bacterium DC413.
The potential of the three strains to hydrolyze plant polysaccharides and the fungal cell wall was analyzed through the annotation of Carbohydrate-Active Enzymes (CAZymes). The complete list of annotated CAZymes is detailed in
Table 4.
P. typographi strains CA3A
T, C2L11, and C2L12B genome sequences annotated 16 GH families; among these, the GH 3 and GH 39 families include β-glucosidases (EC 3.2.1.21), β-xylosidases (EC 3.2.1.37), and α-l-arabinofuranosidase (EC 3.2.1.55). The GH 3 family also include exo-1,3/1,4-β-glucanase (EC 3.2.1) and exo-xyloglucanase (EC 3.2.1.155). The GH 10 and 13 families include α-amylase (EC 3.2.1.1) and α-glucosidase (EC 3.2.1.20), while the GH 15 family includes glucoamylase (EC 3.2.1.3). GH 19 and 23 include chitinases (EC 3.2.1.14) and the GH family 94 includes cellobiose phosphorylase (EC 2.4.1.20), cellodextrin phosphorylase (EC 2.4.1.49), and cellobionic acid phosphorylase (EC 2.4.1.321). In addition, the three genomes were predicted to harbor other CAZymes such as auxiliary activities (AA), carbohydrate esterases (CE), and polysaccharide lyases (PL). The AA 3 family includes cellobiose dehydrogenase (EC 1.1.99.18), the CE 4 family includes acetyl xylan esterase (EC 3.1.1.72), and PL 4 and 26 include rhamnogalacturonan endolyase (EC 4.2.2.23) and rhamnogalacturonan exolyase (EC 4.2.2.24), respectively.
3.3. In Vitro Assays of Catalytic Reactions
In vitro assays of p-nitrophenyl (pNP) substrates for strains CA3AT, C2L11, and C2L12B were used to test a selection of five different enzymatic reactions related to wood polysaccharide hydrolyzation. All three strains showed a weak-positive result for the degradation of α-glucosidase, β-glucosidase, α-xylosidase, β-xylosidase, and cellobiohydrolase.
Petri dishes containing TSA medium that incorporated CMC, xylan, starch, or pectin were inoculated with solutions of P. typographi CA3AT, C2L11, and C2L12B strains at McFarland 7 concentration. The presence of halos on all four different substrates confirmed that all three strains could hydrolyze these substrates.
3.4. Antibiosis Potential
In relation to the antimicrobial potential, the capability of the strains to synthesize siderophores and to inhibit fungal entomopathogens was tested. In vitro analyses for the detection of the predicted synthesis of siderophore molecules were performed on modified M9-CAS-agar medium plates. After inoculation, halos were appreciated within the first 24 h for the three strains.
Antibiosis assays against bark beetle fungal pathogens showed that all three strains of this study are capable of inhibiting fungal pathogens. Nonetheless, whereas strain C2L11 showed a total growth inhibition halo, strains CA3A
T and C2L12B just inhibited the total growth of some of the fungal strains, whereas for other pathogenic fungal strains, just aerial mycelium was inhibited; therefore, it was considered as a partial inhibition (
Figure 4 and
Table 5).
3.5. Other Phenotypic Traits of P. typographi
P. typographi strains CA3AT, C2L11, and C2L12B showed an optimum growth after 48 h incubation at 28 °C on TSA medium, exhibiting the typical iridescent morphology of this genus, although growth was confirmed in the temperature range of 4 to 37 °C. Regarding pH, growth was observed in the range of 6 to 10, but not at pH 4, showing the optimum growth at pH 8. All three strains were able to grow with 0 to 7% NaCl concentration in minimal medium, showing optimal growth with 1% NaCl.
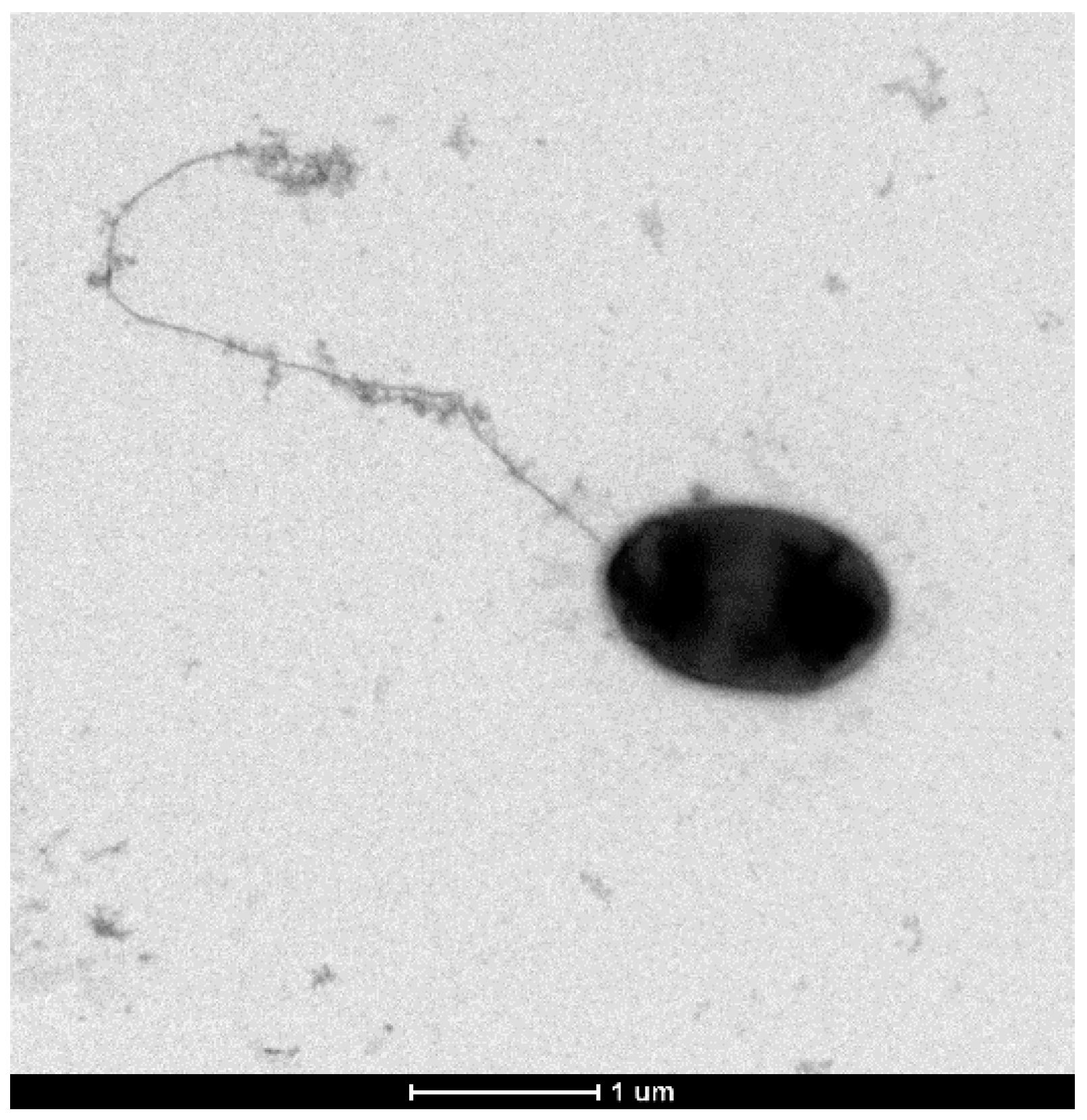
Gram staining showed Gram-negative bacteria and electronic microscopy revealed rod-shaped cells with a polar flagellum (
Figure 5).
After the addition of 30 hydrogen peroxide over the cells of the three strains, the presence of bubbles confirmed catalase activity and blue-stain presence after Kovac’s reactive addition confirmed oxidase activity.
Other phenotypic features of
P. typographi sp. nov. are detailed in the prologue, and those differences with the closely related species are detailed in
Table 6.
4. Discussion
Several different recent researchers have suggested important roles of the bacterial associates in the bark beetle holobiont, but it is a field that is yet to be described in depth. Some of these bacteria are indeed new bacterial species; e.g.,
P. bohemica was firstly isolated from
I. acuminatus and its type strain was described as capable of inhibiting several microbial strains [
13]. In addition, cellulolytic activity was described for the type strain of
P. coleopterorum, first isolated from
Hylesinus fraxini [
54]. Bark beetles cannot digest these compounds, which otherwise would be a potential source of carbon [
4,
15,
54].
In this study, strain CA3A
T was isolated from an adult bark beetle from the species
I. typographus; meanwhile, strains C2L11 and C2L12B were isolated from the second instar larvae of the same host species; all three 16S rRNA gene sequences indicated that these three strains belong to the genus
Pseudomonas, but they showed relatively low similarity to the closest type strains
P. congelans DSM 14939T (97.4),
P. meliae Ogimi 2
T (97.2), and
P. silesiensis A3T (97.2), which suggested that they could belong to a new species within the genus. The phylogenetic analysis of both 16S rRNA and the MLSA of the housekeeping
gyrB, rpoB, and
rpoD gene sequences showed that the three strains locate together in a separated branch from those of the closest
Pseudomonas type strains. ANIb and GGDC values confirmed that all three strains belong to the same species but are distant to the closest type strains, which confirm that they could be considered a new species [
26,
28,
58,
59]. Attending to phenotypical traits, it was observed that unlike the closest strains
P. rhizosphaerae IH5
T, P. alkylphenolica KL28
T, P. entomophila L48
T, and
P. congelans P 538/23
T, all three strains were able to weakly ferment glucose and to degrade urea. In addition, the three new isolates differ from
P. rhizosphaerae IH5
T in the oxidase production and adipate assimilation tests, which are positive for the strains of this study, but negative for
P. rizhosphaerae IH5
T. Moreover, unlike
P. alkylphenolica KL28
T,
P. entomophila L48
T, and
P. congelans P 538/23
T, which are positive for gelatin hydrolysis, N-acetyl-glucosamine and the phenylacetate assimilation test, strains CA3A
T, C2L11, and C2L12B showed negative results. Thus, all these results support the classification of strains CA3A
T, C2L11, and C2L12B as a new species within the genus
Pseudomonas, for which the name
P. typography is proposed.
Apart from that, and in order to evaluate the potential role of these strains on their bark beetle host, an in silico assessment of the genetic potential of the three genome sequences was performed.
Adult bark beetles perforate the external bark to reach the inner bark, where their life cycle will be developed. Firstly, galleries are excavated along the tree and eggs will be laid. Once larvae are born, they excavate their own galleries, develop, and proceed through all the life stages (pupae, young adult, and adults). Once the adult stage is achieved, individuals will leave the tree and seek for a new host to start the cycle again. Thus, almost all of their life cycle develops inside the inner bark, which is composed mainly of cellulose, hemicellulose, and lignin [
1,
3,
4]. Bark beetle cannot digest these compounds by themselves, non-profiting a potential carbon source. It has been proposed that the bark beetle bacteriome might hydrolyze these compounds into simpler sugars, which is potentially profitable for the beetle [
1,
4,
54]. In silico analysis of the genome sequences predicted the presence of CDs related to several CAZYmes families that could be related with this activity, such as GH 3, GH 10, GH 13, GH 15, GH 39, and GH 94, CE 4, AA 3, and PL 4 and PL 26 families. During the hydrolyzation of hemicelluloses (xylans, mannans, mixed linkage β-glucans, and xyloglucans) and cellulose to glucose, the last step will be done by β-glucosidases, which are important enzymes during this process [
60]. In
P. typographi strains, genomes GH 3 and GH 39 were predicted. These families include this enzyme, among other CAZYmes that could be involved in the process, such as β-xylosidases (EC 3.2.1.21), α-
l-arabinofuranosidase (EC 3.2.1.55), exoglucanase (EC 3.2.1.-), and exo-xyloglucanase (EC 3.2.1.155) [
61]. In addition, the CE 4 family includes acetyl xylan esterase (EC 3.1.1.72) [
61]. All these enzymes participate in cellulose and hemicellulose breakdown. The GH 94 and AA 3 families include cellobiose-related enzymes that can also participate in the hydrolysis process [
60]. The GH 10 and 13 families include α-amylase (EC 3.2.1.1) and α-glucosidase (EC 3.2.1.20), the GH 15 family includes glucoamylase (EC 3.2.1.3), and PL 4 and 26 include rhamnogalacturonan endolyase (EC 4.2.2.23) and rhamnogalacturonan exolyase (EC 4.2.2.24), respectively. All these enzymes are related to starch and pectin hydrolysis, which are part of
Picea bark and wood composition [
34,
35,
62,
63]. In order to confirm these enzymatic activities, in vitro assays were performed; pNPs assay confirmed the synthesis of α-glucosidase, β-glucosidase, α-xylosidase, β-xylosidase, and cellobiohydrolase enzymes, and the presence of halos in the plates with CMC, xylan, starch, and pectin as substrates confirmed the hydrolysis capacity of
P. typographi stains. This trait has already been proposed and described for bark beetle bacteriome, including
Pseudomonas strains [
1,
4,
15,
54].
Furthermore, tree’s phloem is known as a sugar-rich source but it is poor in phosphorus, nitrogen, and vitamins, among other important biomolecules and nutrients for the beetle development, for which an important role that could be played by the bacteriome is providing the necessary biomolecules for the beetle, turning non-assimilable mineral compounds to bioassimilable nutrients for the beetle, and more yet to describe [
1,
2,
7,
8,
13,
15]. In this regard, in silico analysis of the genome predicted the potential capacities of these three strains related to metabolic pathways for the reduction of adenyl sulfate (APS) to bioassimilable sulfide, which could suppose a sulfur source for the beetle. In addition, genes related to proteins involved in the biosynthesis of thiamine (vit. B1), riboflavin (vit. B2), pyridoxine (vit. B6), nicotinate (vit. B3), biotine (vit. B8), and folate (vit. B9) were annotated. These are very important for the beetles, since vitamin B is essential for them and they cannot synthetize [
8].
Regarding protection against pathogens, this may occur through the inhibition of pathogens of the beetle itself or to other beetle’s symbionts [
1,
13]. Genome sequences analyses showed that these strains might have antimicrobial potential. In all three genomes, genes belonging to GH 3, GH 19, and GH 23 families, in which chitinase (EC 3.2.1.14) and exo-1,3/1,4-β-Glucanase (EC 3.2.1.-) CAZymes are included, were predicted. Chitinases and glucanases are very important enzymes in fungal cell wall hydrolysis and growth inhibition, which have already been described in
Pseudomonas strains [
64,
65]. In addition, BGCs related to a hypothetical defense role were predicted, such as a biosynthetic bacteriocine cluster and an NRPS-like cluster, similar to AMB from
P. aeruginosa PAO1 (40% similarity) [
66]. Additionally, a BGC similar to crochelin A that could be related to siderophore activity was predicted [
67]. AMB has already been proven as a relatively effective virulence factor against protozoa but only at relatively high concentrations [
68]. Siderophore molecules can inhibit fungal growth, as e.g.,
P. aeruginosa siderophores have already been tested as effective against
Fusarium and
Aspergillus strains [
69]. To confirm these predictions, in vitro siderophore assay was performed, and positive results for the three strains were obtained. Moreover, antibiosis tests using several filamentous fungal entomopathogenic strains of
Ips beetles were accomplished; these tests confirmed the capability of the three strains to inhibit the growth of these fungal entomopathogens. Antimicrobial activity has already been described for other bark beetle isolates, e.g.,
P. bohemica [
13]. Thus, the results obtained in this study support the hypothesis of a potential role of
Pseudomonas associated to bark beetles in the protection of their host. In vivo tests analyzing the beetle survival improvement with the addition of these bacteria to reared beetles should be performed in the future in order to confirm this hypothesis.
In regard to the detoxification of environmental toxic compounds, such as the aromatics emitted by the tree when it is under attack, the analyses of the genome sequences of all three strains predicted routes related to p-hydroxybenzoic acid (PHBA) and salicylate into catechol degradation. Both are important aromatic compounds synthetized by the tree as chemical defenses against pathogens [
70]. Although in vitro and in vivo tests should be performed to confirm the capability of these strains to degrade these aromatics, this role has already been described for
Dendroctonus ponderosae bacteriome [
71].
A NAGGN cluster had potential activity as osmotic stress resistance, as described for
P. aeruginosa [
72]. Finally, a carotenoid BGC was also predicted; generally, these pigments cannot be synthesized by animals, and it has already been described that some insect symbionts can synthesize them [
73,
74].
5. Conclusions
In this study, three closely related strains belonging to the genus Pseudomonas were isolated from individuals at different stages of the I. typographus life cycle. Genomes sequences of these three strains show their potential role in nutrient provisioning, protection against pathogens, and toxic compounds degradation. In vitro tests confirmed the capability of strains CA3AT, C2L11, and C2L12B to inhibit fungal entomopathogens of the beetle, as well as their ability to hydrolyze cellulose, xylan, starch, and pectin. A polyphasic taxonomic approach indicated that these strains belong to a new species within the genus Pseudomonas, for which the name P. typographi has been proposed.
Although further analyses are needed to confirm that the predicted capabilities occur within I. typographus holobiont and benefit the host, we can conclude that the new species P. typographi sp. nov. has genetic potential to aid in its host ecology, and the isolation of members of this new species from two different stages of the life cycle of the beetle also supports the possible relevance of this species for the bark beetle holobiont.
Description of Pseudomonas typographi sp. nov.
Pseudomonas typographi (ty.po.gra.phi. M.L. adj. related to Ips typographus, the bark beetle where the type strain was isolated from). Cells are Gram-negative, rod shaped, with approximately 1 µm length and 0.5–0.6 µm width. On TSA medium, the optimal growth temperature is 28 °C, although good growth happens in the range of 4 to 37 °C; on TSB medium, it tolerates NaCl from 0 to 4, with an optimal growth with 1 NaCl; the pH growth range is 6 to 10, with an optimum at 8. Cells are catalase and oxidase positive. Results in the API 20 NE system are positive in the reduction of nitrates, D-glucose fermentation, arginine DiHydrolase, urease, assimilation of D-glucose, L-arabinose, D-mannose, D-mannitol, potassium gluconate, caprate, adipate, and trisodium citrate were also positive, whereas indole production, gelatin hydrolysis, β-galactosidase, N-acetyl-glucosamine, D-maltose, and phenylacetate were negative. In API 50CH, the type strain produced acid from glycerol, erythritol, l-arabinose, d-ribose, d-xylose, d-galactose, d-glucose, d-fructose, d-mannose, l-rhamnose, inositol, d-sorbitol, d-maltose, gentiobiose, d-turanose, d-fucose, and d-arabitol.
The type strain, CA3AT (= CECT 30101T = LMG 31781T), was isolated from a bark beetle from the species Ips typographus in Czech Republic. The DNA G + C of the type strain is 62.1 mol.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}