Comparative Mitogenomic Analysis of Two Cuckoo Bees (Apoidea: Anthophila: Megachilidae) with Phylogenetic Implications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. Sequencing and Assembly
2.3. Bioinformatic Analysis
2.4. Phylogeny Analysis
3. Results and Discussion
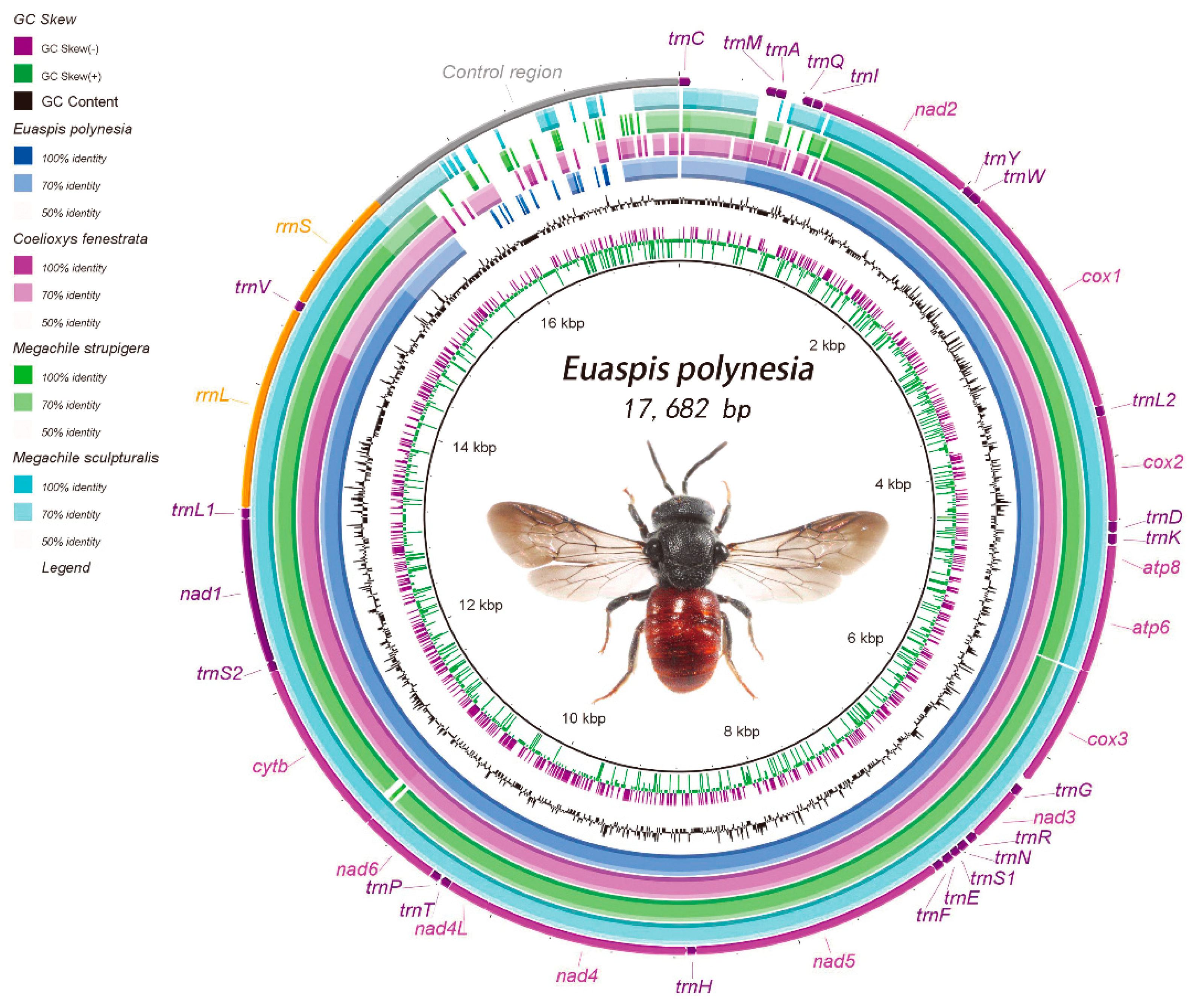
3.1. General Features of the Mitogenome of C. fenestrata and E. polynesia
3.2. Genome Structure
3.2.1. Protein-Coding Genes
3.2.2. Transfer RNA Genes
3.2.3. Ribosomal RNA Genes
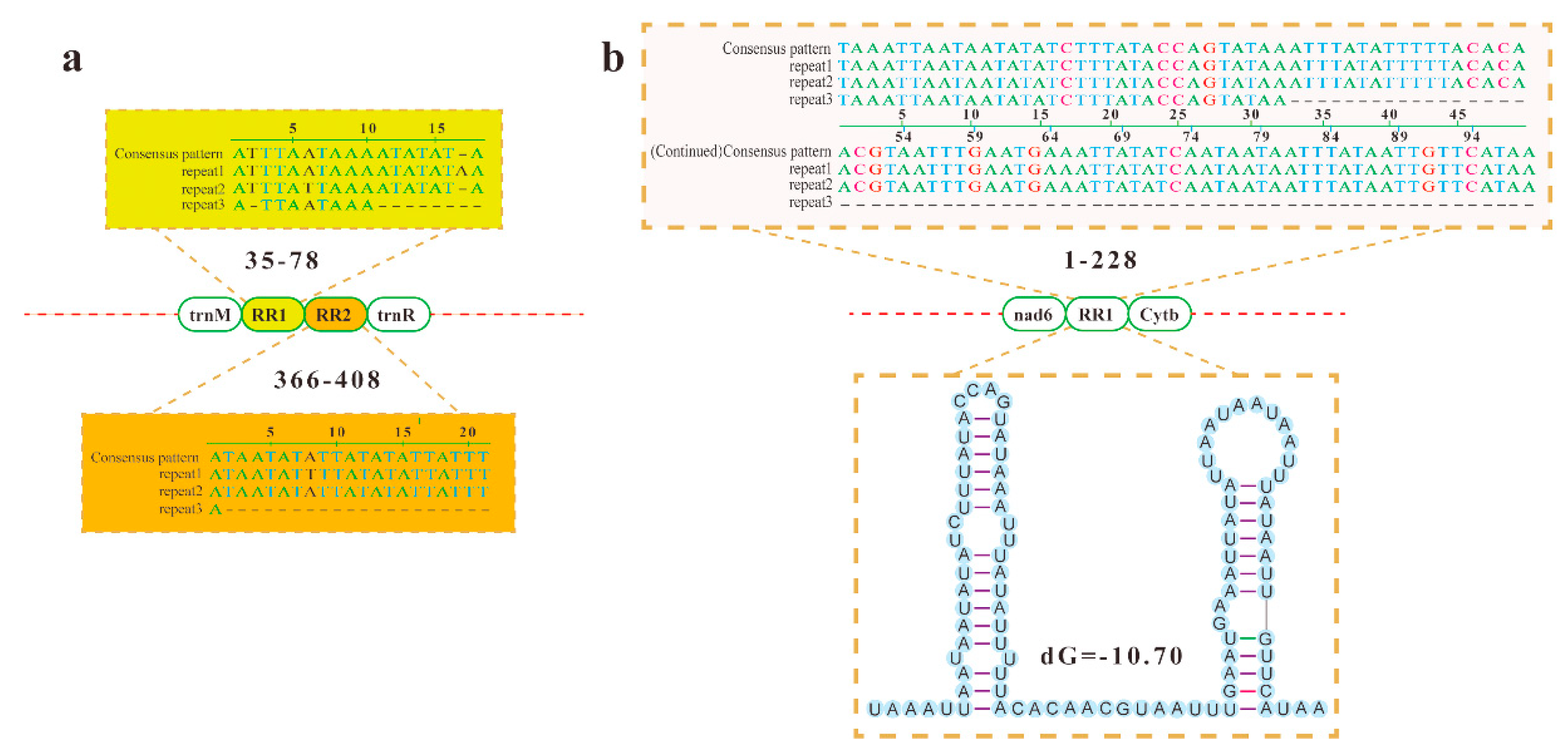
3.2.4. Noncoding Regions
3.2.5. Gene Rearrangement
3.3. Nucleotide Diversity
3.4. Phylogenetic Analysis
3.4.1. Substitution Saturation Tests
3.4.2. Topology Consistency Analysis
3.4.3. Phylogenetic Relationship
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michener, C.D. The Bees of the World, 2nd ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2007; pp. 434–543. ISBN 9780801885730. [Google Scholar]
- Danforth, B.N.; Cardinal, S.; Praz, C.; Almeida, E.A.B.; Michez, D. The Impact of Molecular Data on Our Understanding of Bee Phylogeny and Evolution. Annu. Rev. Entomol. 2013, 58, 57–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascher, J.S.; Pickering, J. Discover Life Bee Species Guide and World Checklist (Hymenoptera: Apoidea: Anthophila). Draft-50. 2018. Available online: http://www.discoverlife.org/mp/20q?guide=Apoidea_species (accessed on 31 March 2018).
- Litman, J.R.; Danforth, B.N.; Eardley, C.D.; Praz, C.J. Why do leafcutter bees cut leaves? New insights into the early evolution of bees. Proc. R. Soc. B. 2011, 278, 3593–3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sabino, W.O.; Antonini, Y. Nest Architecture, Life Cycle, and Natural Enemies of the Neotropical Leafcutting Bee Megachile (Moureapis) maculata (Hymenoptera: Megachilidae) in a Montane Forest. Apidologie 2017, 48, 450–460. [Google Scholar]
- Gonzalez, V.H.; Griswold, T.; Praz, C.J.; Danforth, B.N. Phylogeny of the Bee Family Megachilidae (Hymenoptera: Apoidea) Based on Adult Morphology. Syst. Entomol. 2012, 37, 261–286. [Google Scholar] [CrossRef] [Green Version]
- Pitts-Singer, T.L.; Cane, J.H. The Alfalfa Leaf-Cutting Bee, Megachile rotundata: The World’s Most Intensively Managed Solitary Bee. Annu. Rev. Entomol. 2011, 56, 221–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, M.E.; Favi, F.D. Flower Phenology and Pollen Choice of Osmia lignaria (Hymenoptera: Megachilidae) in Central Virginia. Environ. Entomol. 2005, 34, 1593–1605. [Google Scholar] [CrossRef]
- Vicens, N.; Bosch, J. Weather-Dependent Pollinator Activity in an Apple Orchard, with Special Reference to Osmia cornuta and Apis mellifera (Hymenoptera: Megachilidae and Apidae). Environ. Entomol. 2000, 29, 413–420. [Google Scholar] [CrossRef]
- Soh, E.J.Y.; Soh, Z.W.W.; Chui, S.X.; Ascher, J.S. The Bee Tribe Anthidiini in Singapore (Anthophila: Megachilidae: Anthidiini) with Notes on the Regional Fauna. Nat. Singap. 2016, 9, 49–62. [Google Scholar]
- Baker, D.B. A Review of Asian Species of Genus Euaspis Gerstäcker (Hymenoptera: Apoidea: Megachilidae). Zool. Meded. 1995, 69, 281–302. [Google Scholar]
- Nadimi, A.; Talebi, A.A.; Fathipour, Y. A Preliminary Study of the Cleptoparasitic Bees of the Genus Coelioxys (Hymenoptera: Megachilidae) in Northern Iran, with Six New Records. J. Crop Prot. 2013, 2, 271–283. [Google Scholar]
- Filho, L.C.; Garófalo, C.A. Nesting Biology of Megachile (Chrysosarus) guaranitica and High Mortality Caused by Its Cleptoparasite Coelioxys bertonii (Hymenoptera: Megachilidae) in Brazil. Austral Entomol. 2015, 55, 25–31. [Google Scholar] [CrossRef]
- Roig-Alsina, A.; Michener, C.D. Studies of the Phylogeny and Classification of Long-Tongued Bees (Hymenoptera: Apoidea). Kans. Univ. Sci. Bull. 1993, 55, 124–162. [Google Scholar]
- Wu, Y.R. Fauna Sinica: Insecta, Volume 44: Hymenoptera: Megachilidae; Science Press: Beijing, China, 2006; pp. 2–3. ISBN 7-03-016332-X. [Google Scholar]
- Engel, M.S. A Monograph of the Baltic Amber Bees and Evolution of the Apoidea (Hymenoptera). Bull. Am. Mus. Nat. Hist. 2001, 259, 1–192. [Google Scholar] [CrossRef]
- Praz, C.J.; Müller, A.; Danforth, B.N.; Griswold, T.L.; Widmer, A.; Dorn, S. Phylogeny and Biogeography of Bees of the Tribe Osmiini (Hymenoptera: Megachilidae). Mol. Phylogenet. Evol. 2008, 49, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahnt, B.; Gerth, M.; Paxton, R.J.; Bleidorn, C.; Husemann, M. The Complete Mitochondrial Genome of the Endemic and Highly Specialized South African Bee Species Rediviva intermixta (Hymenoptera: Melittidae), with a Comparison with Other Bee Mitogenomes. Biol. J. Linn. Soc. 2015, 116, 940–953. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Gibson, T.; Dowton, M. Higher-Level Phylogeny of the Hymenoptera Inferred from Mitochondrial Genomes. Mol. Phylogenet. Evol. 2015, 84, 34–43. [Google Scholar] [CrossRef]
- Song, S.N.; Tang, P.; Wei, S.J.; Chen, X.X. Comparative and Phylogenetic Analysis of the Mitochondrial Genomes in Basal Hymenopterans. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, L.X.; Xiang, D.B.; Wan, Y.; Wu, Q.; Huang, W.L.; Zhao, G. The Complete Mitochondrial Genomes of Two Model Ectomycorrhizal Fungi (Laccaria): Features, Intron Dynamics and Phylogenetic Implications. Int. J. Biol. Macromol. 2020, 145, 974–984. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the Phylogeny of Holometabola with Mitochondrial Genome Data and Bayesian Site-Heterogeneous Mixture Models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Song, F.; Zhao, Y.; Wilson, J.J.; Cai, W. Higher-Level Phylogeny and Evolutionary History of Pentatomomorpha (Hemiptera: Heteroptera) Inferred from Mitochondrial Genome Sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Jin, Y.; Cai, W.; Sota, T.; Li, H. Mitochondrial Genomics Reveals Shared Phylogeographic Patterns and Demographic History among Three Periodical Cicada Species Groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Cameron, S.L.; Dowavic, J.I.; Austin, A.D.; Whiting, M.F. Characterization of 67 Mitochondrial tRNA Gene Rearrangements in the Hymenoptera Suggests that Mitochondrial tRNA Gene Position Is Selectively Neutral. Mol. Biol. Evol. 2009, 26, 1607–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Li, H.; Shao, R.; Shi, A.; Bai, X.; Zheng, X.; Heiss, E.; Cai, W. Rearrangement of Mitochondrial tRNA Genes in Flat Bugs (Hemiptera: Aradidae). Sci. Rep. 2016, 6, 25725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Su, T.J.; He, B.; Gu, P.; Huang, D.Y.; Zhu, C.D. Sequencing and Characterization of the Megachile sculpturalis (Hymenoptera: Megachilidae) Mitochondrial Genome. Mitochondrial DNA Part A 2015, 28, 309–311. [Google Scholar] [CrossRef]
- Huang, D.Y.; Su, T.J.; He, B.; Gu, P.; Liang, A.P.; Zhu, C.D. Sequencing and Characterization of the Megachile strupigera (Hymenoptera: Megachilidae) Mitochondrial Genome. Mitochondrial DNA Part B 2016, 1, 309–311. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.Y.; Cao, L.J.; Tang, P.; van Achterberg, K.; Hoffmann, A.A.; Chen, H.Y.; Chen, X.X.; Wei, S.J. Gene Arrangement and Sequence of Mitochondrial Genomes Yield Insights Into the Phylogeny and Evolution of Bees and Sphecid Wasps (Hymenoptera: Apoidea). Mol. Phylogenet. Evol. 2018, 124, 1–9. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing Mitochondrial Genomes Directly from Genomic Next-Generation Sequencing Reads—A Baiting and Iterative Mapping Approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARWEN: A Program to Detect tRNA Genes in Metazoan Mitochondrial Nucleotide Sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE Online: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Golosova, O.; Sukhomlinov, D.; Tiunov, A.; Prosperi, M. Flexible Design of Multiple Metagenomics Classification Pipelines with UGENE. Bioinformatics 2019, 35, 1963–1965. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the Nuclear (18S, 5.8S, 28S and 5S) and Mitochondrial (12S and 16S) rRNA Genes of Apis mellifera (Insecta: Hymenoptera): Structure, Organization, and Retrotransposable Elements. Insect Mol. Biol. 2006, 15, 657–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.J.; Tang, P.; Zheng, L.H.; Shi, M.; Chen, X.X. The Complete Mitochondrial Genome of Evania appendigaster (Hymenoptera: Evaniidae) Has Low A + T Content and a Long Intergenic Spacer between atp8 and atp6. Mol. Biol. Rep. 2010, 37, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.J.; He, B.; Li, K.; Liang, A.P. Comparative Analysis of the Mitochondrial Genomes of Oriental Spittlebug Trible Cosmoscartini: Insights Into the Relationships among Closely Related Taxa. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- He, B.; Su, T.J.; Niu, Z.Q.; Zhou, Z.Y.; Gu, Z.Y.; Huang, D.Y. Characterization of Mitochondrial Genomes of Three Andrena Bees (Apoidea: Andrenidae) and Insights Into the Phylogenetics. Int. J. Biol. Macromol. 2019, 127, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The Comparative RNA Web (CRW) Site: An Online Database of Comparative Sequence and Structure Information for Ribosomal, Intron, and Other RNAs. BMC Bioinform. 2002, 3, 2. [Google Scholar]
- Reuter, J.S.; Mathews, D.H. RNA Structure: Software for RNA Secondary Structure Prediction and Analysis. BMC Bioinform. 2010, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.F.; Petty, N.K.; Zakour, N.L.B.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An Integrated and Scalable Desktop Platform for Streamlined Molecular Sequence Data Management and Evolutionary Phylogenetics Studies. Mol. Ecol. Res. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A Web Server for Microsatellite Prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring Genomic Rearrangements Based on Common Intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarisation in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.H.; Huang, D.Y. The Complete Mitogenome of Habropoda rodoszkowskii (Hymenoptera: Apidae) and Phylogenetic Analysis. Mitochondrial DNA Part B 2020, 5, 2350–2351. [Google Scholar] [CrossRef]
- He, B.; Su, T.J.; Wu, Y.P.; Xu, J.S.; Huang, D.Y. Phylogenetic Analysis of the Mitochondrial Genomes in Bees (Hymenoptera: Apoidea: Anthophila). PLoS ONE 2018, 13, e0202187. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA Punctuation Model of RNA Processing in Human Mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Li, H.Y.; Lu, H.H.; Huang, S.G.; Fan, X.D.; Luo, A.C.; Huang, D.Y. The Complete Mitogenome of Nomia chalybeata (Hymenoptera: Halictidae) and Phylogenetic Analysis. Mitochondrial DNA Part B 2020, 5, 2850–2851. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A Novel Type of RNA Editing Occurs in the Mitochondrial tRNAs of the Centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, S.L.; Whiting, M.F. The Complete Mitochondrial Genome of the Tobacco Hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an Examination of Mitochondrial Gene Variability within Butterflies and Moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.L.; Huang, D.Y.; Sun, X.Y.; Hao, J.S.; Cheng, C.H.; Zhang, W.; Yang, Q. Complete Mitochondrial Genome Sequence of Cheirotonus jansoni (Coleoptera: Scarabaeidae). Genet. Mol. Res. 2014, 13, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Su, T.J.; Liang, A.P. Characterization of the Complete Mitochondrial Genome of Phymatostetha huangshanensis (Hemiptera: Cercopidae) and Phylogenetic Analysis. Int. J. Biol. Macromol. 2018, 119, 60–69. [Google Scholar] [CrossRef]
- Park, J.K.; Kim, K.H.; Kang, S.; Kim, W.; Eom, K.S.; Littlewood, D.T. A Common Origin of Complex Life Cycles in Parasitic Flatworms: Evidence from the Complete Mitochondrial Genome of Microcotyle sebastis (Monogenea: Platyhelminthes). BMC Evol. Biol. 2007, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, W.X.; Zou, H.; Wu, S.G.; Li, M.; Jakovlić, I.; Zhang, J.; Chen, R.; Wang, G.T. Mitochondrial Genomes and 28S rDNA Contradict the Proposed Obsoletion of the Order Tetraonchidea (Platyhelminthes: Monogenea). Int. J. Biol. Macromol. 2019, 143, 891–901. [Google Scholar] [CrossRef]
- Le, T.H.; Blair, D.; McManus, D.P. Mitochondrial Genomes of Parasitic Flatworms. Trends Parasitol. 2002, 18, 206–213. [Google Scholar] [CrossRef]
- Fumagalli, L.; Taberlet, P.; Favre, L.; Hausser, J. Origin and Evolution of Homologous Repeated Sequences in the Mitochondrial DNA Control Region of Shrews. Mol. Biol. Evol. 1996, 13, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.Y.; Li, J.; Geng, S.; Yang, J.; Zhang, X.; An, Y.X.; Li, C.; Cui, H.R.; Li, X.Y.; Wang, Y.Y. The First Mitochondrial Genome for Phaudidae (Lepidoptera) with Phylogenetic Analyses of Zygaenoidea. Int. J. Biol. Macromol. 2020, 149, 951–961. [Google Scholar] [CrossRef]
- Dowton, M.; Austin, A.D. Evolutionary Dynamics of a Mitochondrial Rearrangement “Hot Spot” in the Hymenoptera. Mol. Biol. Evol. 1999, 16, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowton, M.; Castro, L.R.; Campbell, S.L.; Bargon, S.D.; Austin, A.D. Frequent Mitochondrial Gene Rearrangements at the Hymenopteran nad3-nad5 Junction. J. Mol. Evol. 2003, 56, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, P.Y.; Xue, X.F.; Hua, H.Q.; Li, Y.X.; Zhang, F.; Wei, S.J. Extensive Gene Rearrangements in the Mitochondrial Genomes of Two Egg Parasitoids, Trichogramma japonicum and Trichogramma ostriniae (Hymenoptera: Chalcidoidea: Trichogrammatidae). Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pureum, N.; Wook, J.K.; Jun-Ho, S.; Inkyu, P.; Goya, C.; Byeong, C.M. Rapid and Simple Species Identification of Cicada Exuviae Using COI-Based SCAR Assay. Insects 2020, 11, 168. [Google Scholar]
- Elaine, F.; Alexandre, R.Z.; Paulo, C.R.; João, P.; Naldi, S.; Rute, B.; Benjamin, P.O.; Maria, C.A. Conserved Numts Mask a Highly Divergent Mitochondrial-COI Gene in a Species Complex of Australian Stingless Bees Tetragonula (Hymenoptera: Apidae). Mitochondrial DNA Part A 2019, 30, 806–817. [Google Scholar]
- Goldstein, D.B.; Linares, A.R.; Cavalli-Sforza, L.L.; Feldman, M.W. An Evaluation of Genetic Distances for Use with Microsatellite Loci. Genetics 1995, 139, 463–471. [Google Scholar]
- Cameron, S.L.; Johnson, K.P.; Whiting, M.F. The Mitochondrial Genome of the Screamer Louse Bothriometopus (Phthiraptera: Ischnocera): Effects of Extensive Gene Rearrangements on the Evolution of the Genome. J. Mol. Evol. 2007, 65, 589–604. [Google Scholar] [CrossRef]
- Yang, X.S.; Cameron, S.L.; Lees, D.C.; Xue, D.Y.; Han, H.X. A Mitochondrial Genome Phylogeny of Owlet Moths (Lepidoptera: Noctuoidea), and Examination of the Utility of Mitochondrial Genomes for Lepidopteran Phylogenetics. Mol. Phylogenet. Evol. 2015, 85, 230–237. [Google Scholar] [CrossRef]
- Cameron, S.L.; Lo, N.; Bourguignon, T.; Svenson, G.J.; Evans, T.A. A Mitochondrial Genome Phylogeny of Termites (Blattodea: Termitoidae): Robust Support for Interfamilial Relationships and Molecular Synapomorphies Define Major Clades. Mol. Phylogenet. Evol. 2012, 65, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Nelson, L.A.; Lambkin, C.L.; Batterham, P.J.; Wallman, J.F.; Dowton, M.; Whiting, M.F.; Yeatesa, D.K.; Camerong, S.L. Beyond Barcoding: A Mitochondrial Genomics Approach to Molecular Phylogenetics and Diagnostics of Blowflies (Diptera: Calliphoridae). Gene 2012, 511, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.S.; Xue, D.Y.; Han, H.X. The Complete Mitochondrial Genome of Biston panterinaria (Lepidoptera: Geometridae), with Phylogenetic Utility of Mitochondrial Genome in the Lepidoptera. Gene 2013, 515, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Dowton, M. Complete Mitochondrial Genomes of Ceratobaeus sp. and Idris sp. (Hymenoptera: Scelionidae): Shared Gene Rearrangements as Potential Phylogenetic Markers at the Tribal Level. Mol. Biol. Rep. 2014, 41, 6419–6427. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, M.N.; Korkmaz, E.M. Comparative Mitogenomics of Hymenoptera Reveals Evolutionary Differences in Structure and Composition. Int. J. Biol. Macromol. 2019, 144, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.G.; Litman, J.R.; Danforth, B.N. Rooting Phylogenies Using Gene Duplications: An Empirical Example from the Bees (Apoidea). Mol. Phylogenet. Evol. 2011, 60, 295–304. [Google Scholar] [CrossRef]
- Dellicour, S.; Lecocq, T.; Kuhlmann, M.; Mardulyn, P.; Michez, D. Molecular Phylogeny, Biogeography, and Host Plant Shifts in the Bee Genus Melitta (Hymenoptera: Anthophila). Mol. Phylogenet. Evol. 2014, 70, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Debevec, A.H.; Cardinal, S.; Danforth, B.N. Identifying the Sister Group to the Bees: A Molecular Phylogeny of Aculeata with an Emphasison the Superfamily Apoidea. Zool. Scr. 2012, 41, 527–535. [Google Scholar] [CrossRef]
- Hedtke, S.M.; Patiny, S.; Danforth, B.N. The Bee Tree of Life: A Supermatrix Approach to Apoid Phylogeny and Biogeography. BMC Evol. Biol. 2013, 13, 138. [Google Scholar] [CrossRef] [Green Version]
- Peters, R.S.; Krogmann, L.; Mayer, C.; Donath, A.; Gunkel, S.; Meusemann, K.; Kozlov, A.; Podsiadlowski, L.; Petersen, M.; Lanfear, R.; et al. Evolutionary History of the Hymenoptera. Curr. Biol. 2017, 27, 1013–1018. [Google Scholar] [CrossRef] [Green Version]
- Branstetter, M.G.; Danforth, B.N.; Pitts, J.P.; Faircloth, B.C.; Ward, P.S.; Buffington, M.L.; Gates, M.W.; Kula, R.R.; Brady, S.G. Phylogenomic Insights Into the Evolution of Stinging Wasps and the Origins of Ants and Bees. Curr. Biol. 2017, 27, 1019–1025. [Google Scholar] [CrossRef] [Green Version]
- Sann, M.; Niehuis, O.; Peters, R.S.; Mayer, C.; Kozlov, A.; Podsiadlowski, L.; Bank, S.; Meusemann, K.; Misof, B.; Bleidorn, C.; et al. Phylogenomic Analysis of Apoidea Sheds New Light on the Sister Group of Bees. BMC Evol. Biol. 2018, 18, 71. [Google Scholar] [CrossRef] [Green Version]
- Litman, J.R.; Praz, C.J.; Danforth, B.N.; Griswold, T.L.; Cardinal, S. Origins, evolution, and diversification of cleptoparasitic lineages in long-tongued bees. Evolution 2013, 67, 2982–2998. [Google Scholar] [CrossRef] [PubMed]
- Litman, J.R.; Griswold, T.; Danforth, B.N. Phylogenetic systematics and a revised generic classification of anthidiine bees (Hymenoptera: Megachilidae). Mol. Phylogenet. Evol. 2016, 100, 183–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; He, B.; Hao, Y.; Zhou, Z.; Su, C.; Huang, D. Comparative Mitogenomic Analysis of Two Cuckoo Bees (Apoidea: Anthophila: Megachilidae) with Phylogenetic Implications. Insects 2021, 12, 29. https://doi.org/10.3390/insects12010029
Lu H, He B, Hao Y, Zhou Z, Su C, Huang D. Comparative Mitogenomic Analysis of Two Cuckoo Bees (Apoidea: Anthophila: Megachilidae) with Phylogenetic Implications. Insects. 2021; 12(1):29. https://doi.org/10.3390/insects12010029
Chicago/Turabian StyleLu, Huanhuan, Bo He, Youjin Hao, Zeyang Zhou, Chengyong Su, and Dunyuan Huang. 2021. "Comparative Mitogenomic Analysis of Two Cuckoo Bees (Apoidea: Anthophila: Megachilidae) with Phylogenetic Implications" Insects 12, no. 1: 29. https://doi.org/10.3390/insects12010029
APA StyleLu, H., He, B., Hao, Y., Zhou, Z., Su, C., & Huang, D. (2021). Comparative Mitogenomic Analysis of Two Cuckoo Bees (Apoidea: Anthophila: Megachilidae) with Phylogenetic Implications. Insects, 12(1), 29. https://doi.org/10.3390/insects12010029