
Diaphorina citri Genome Possesses a Complete Melatonin Biosynthesis Pathway Differentially Expressed under the Influence of the Phytopathogenic Bacterium, Candidatus Liberibacter asiaticus



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Analysis
2.1.1. Protein–Protein BLAST (BLASTp) Analysis
2.1.2. Evolutionary Analysis by Maximum Likelihood Method
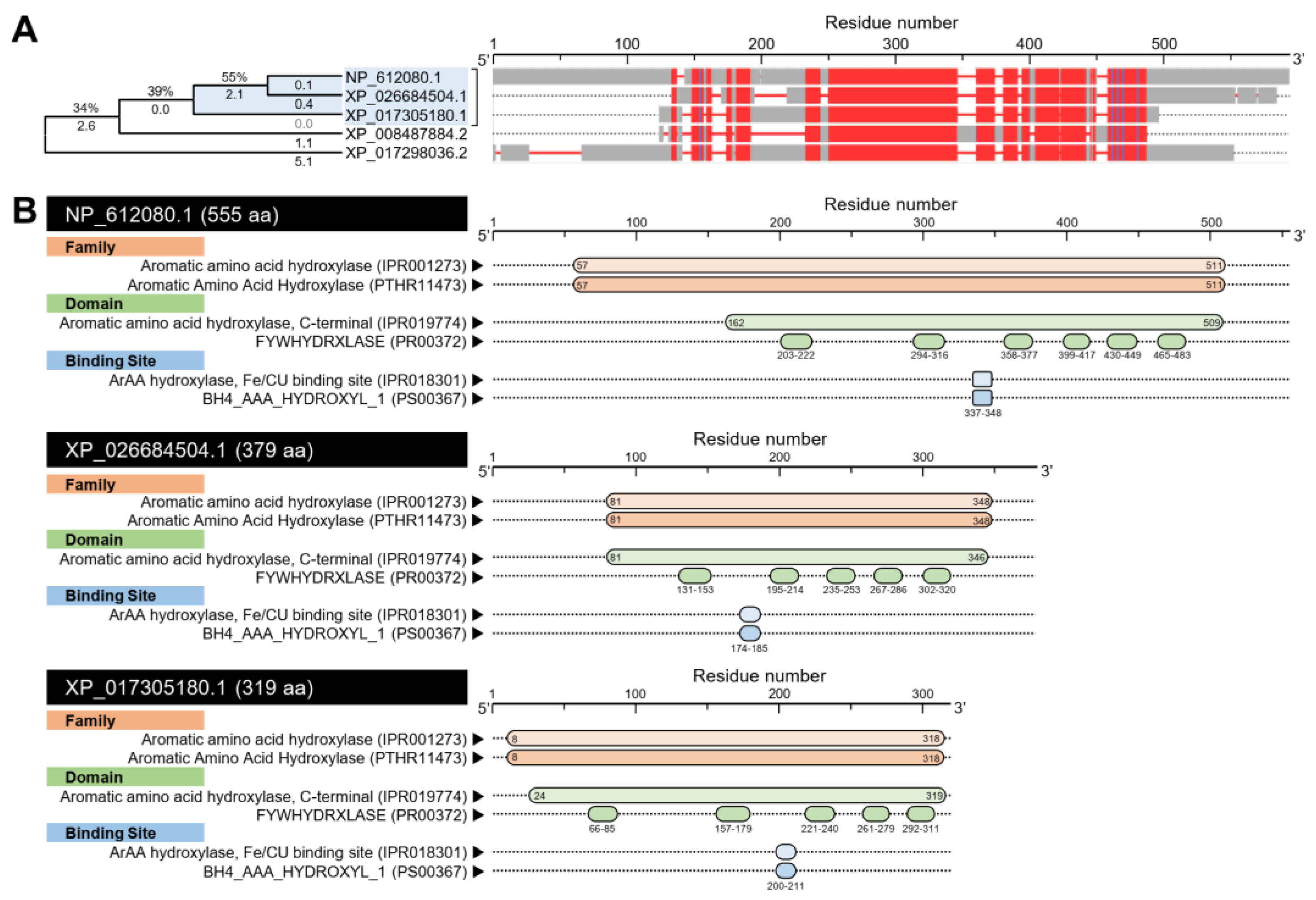
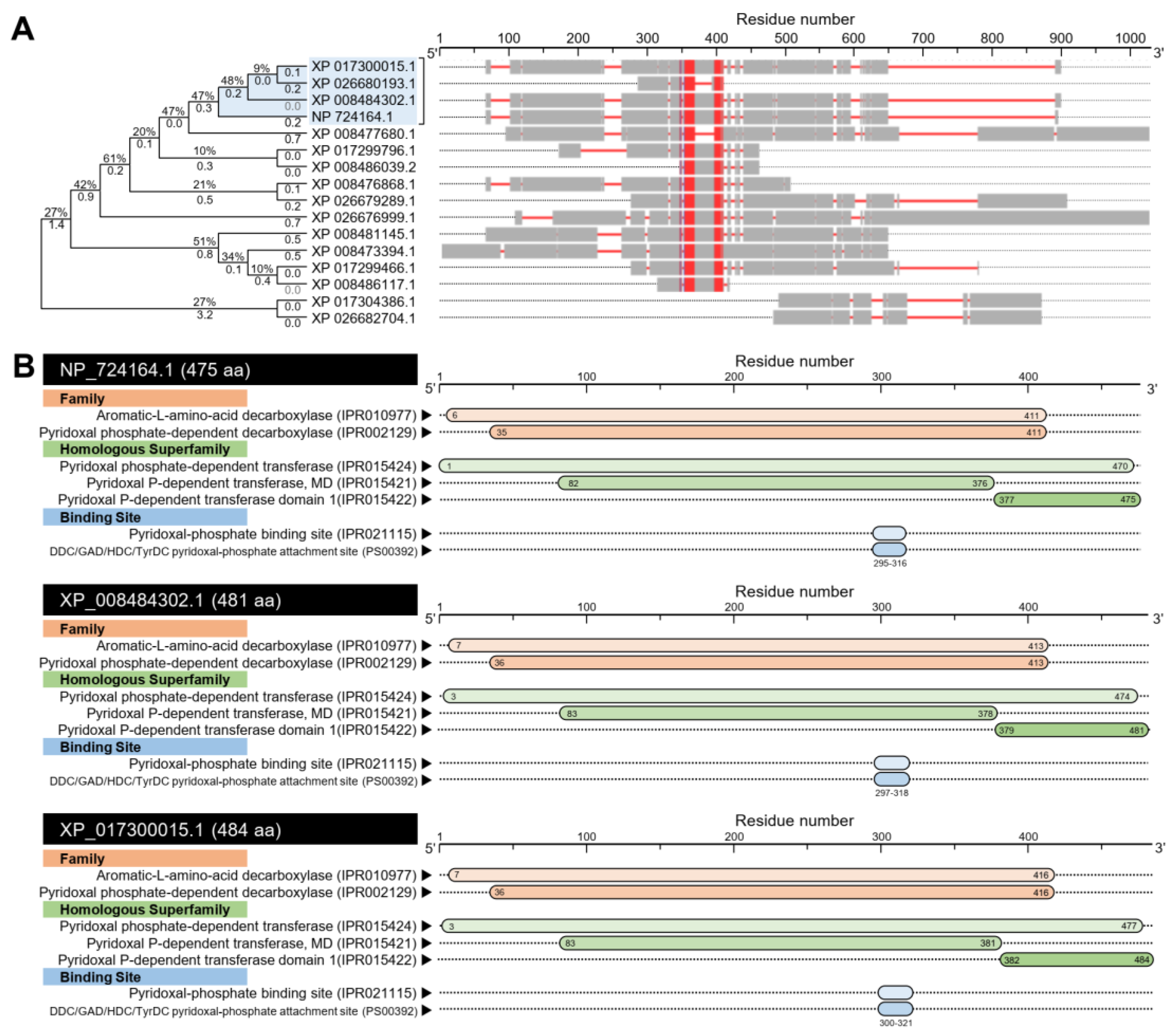
2.1.3. Multiple Sequence Alignment Analysis
2.1.4. Conserved Domains and Theoretical pI/Mw
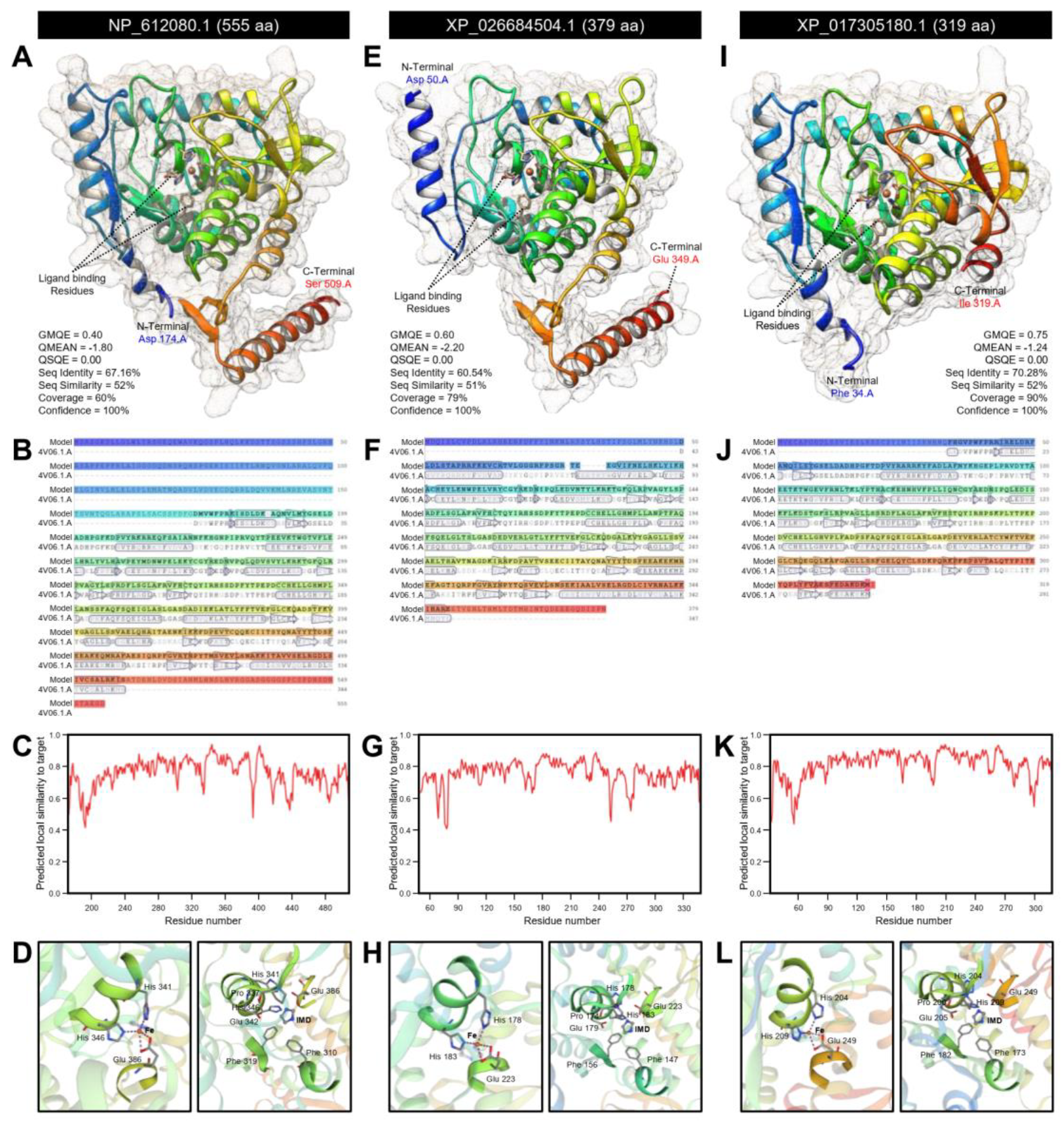
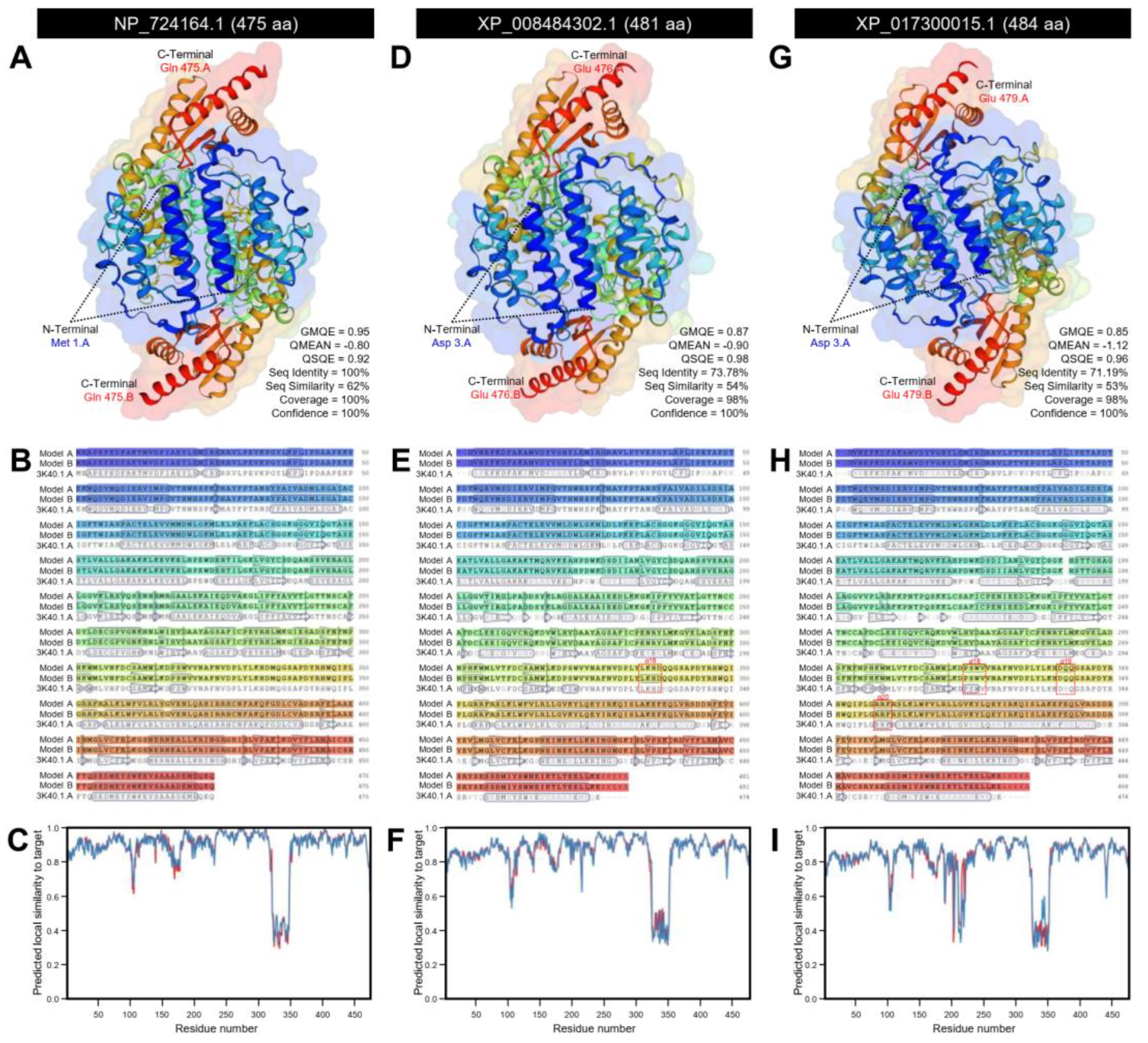
2.1.5. Three-Dimensional (3D) Structure Modeling and RNA Secondary Structure
2.2. Rearing of Healthy and Ca. L. asiaticus-Infected D. citri Colonies
2.3. Treatment with Exogenous Melatonin
2.4. Gene Expression Analysis Using Quantitative Real-Time PCR (RT-PCR)
2.5. Statistical Analysis
3. Results
3.1. D. citri Genome Possesses a Putative Melatonin Biosynthetic Pathway
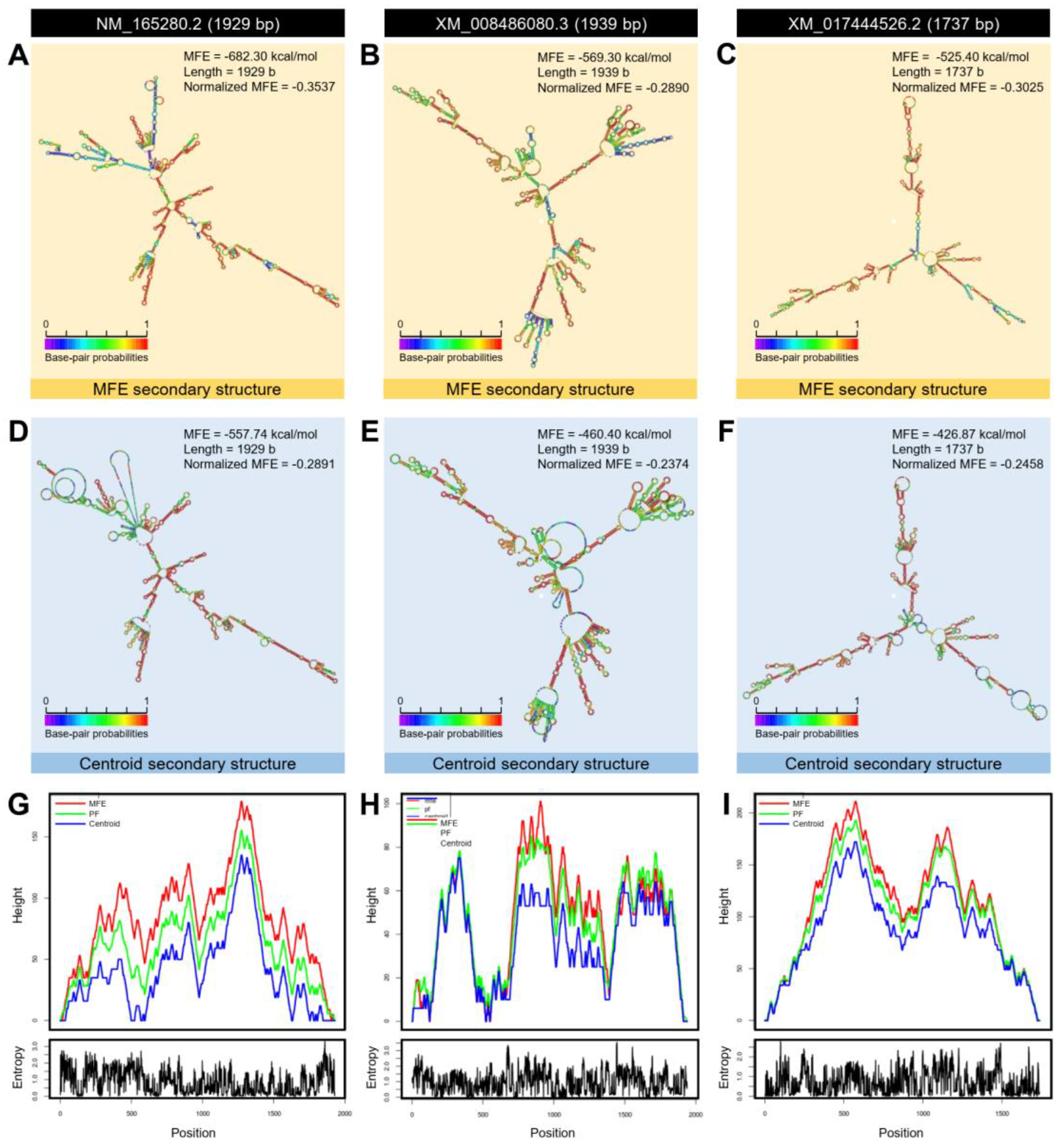
3.1.1. D. citri Genome Encodes for two Putative Tryptophan 5-hydroxylase (DcT5H)
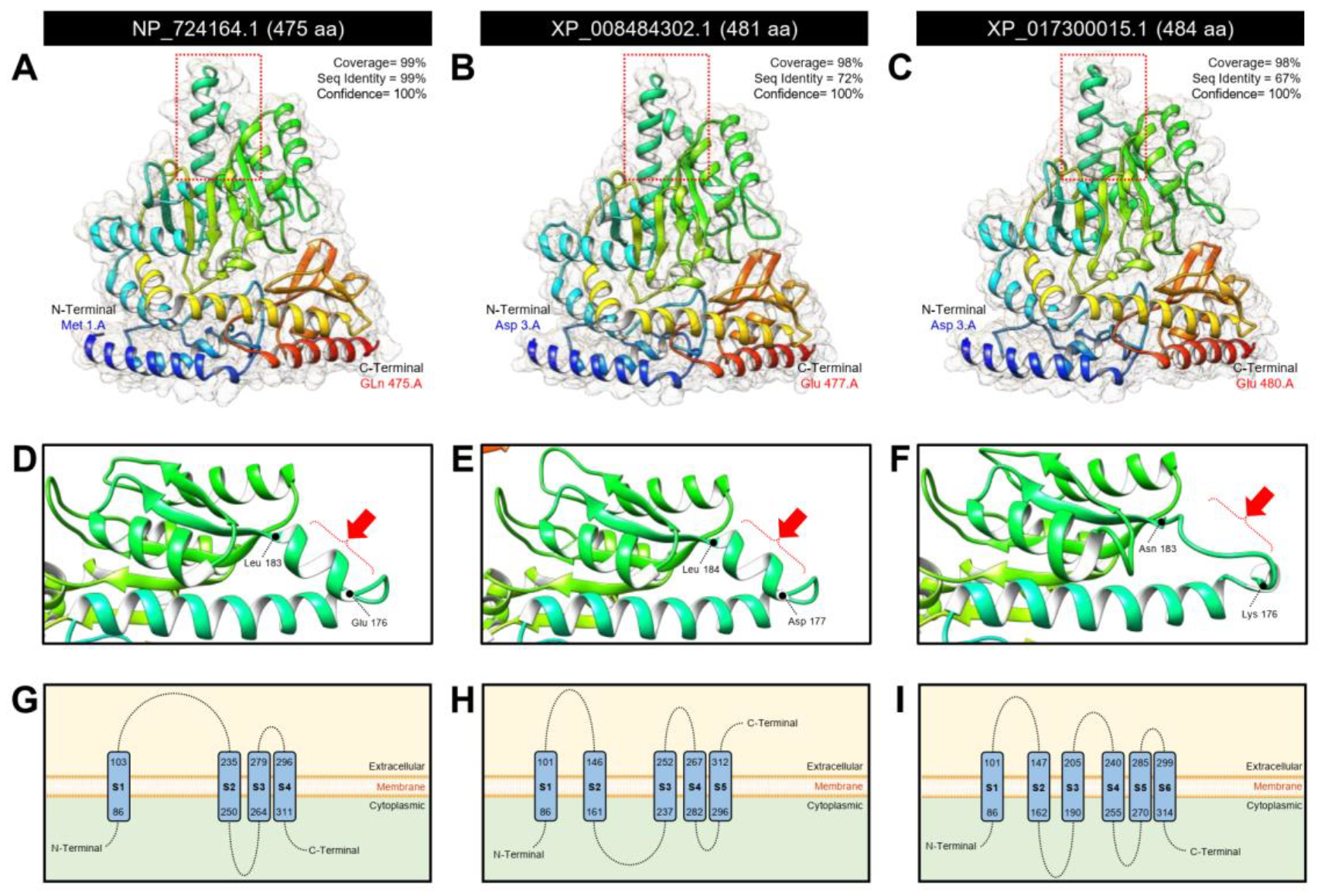
3.1.2. D. citri Genome Encodes for a Putative Aromatic Amino Acid Decarboxylase (DcAADC)
3.1.3. D. citri Genome Possesses Two Putative Arylalkylamine N-acetyltransferase Genes (DcAANAT)
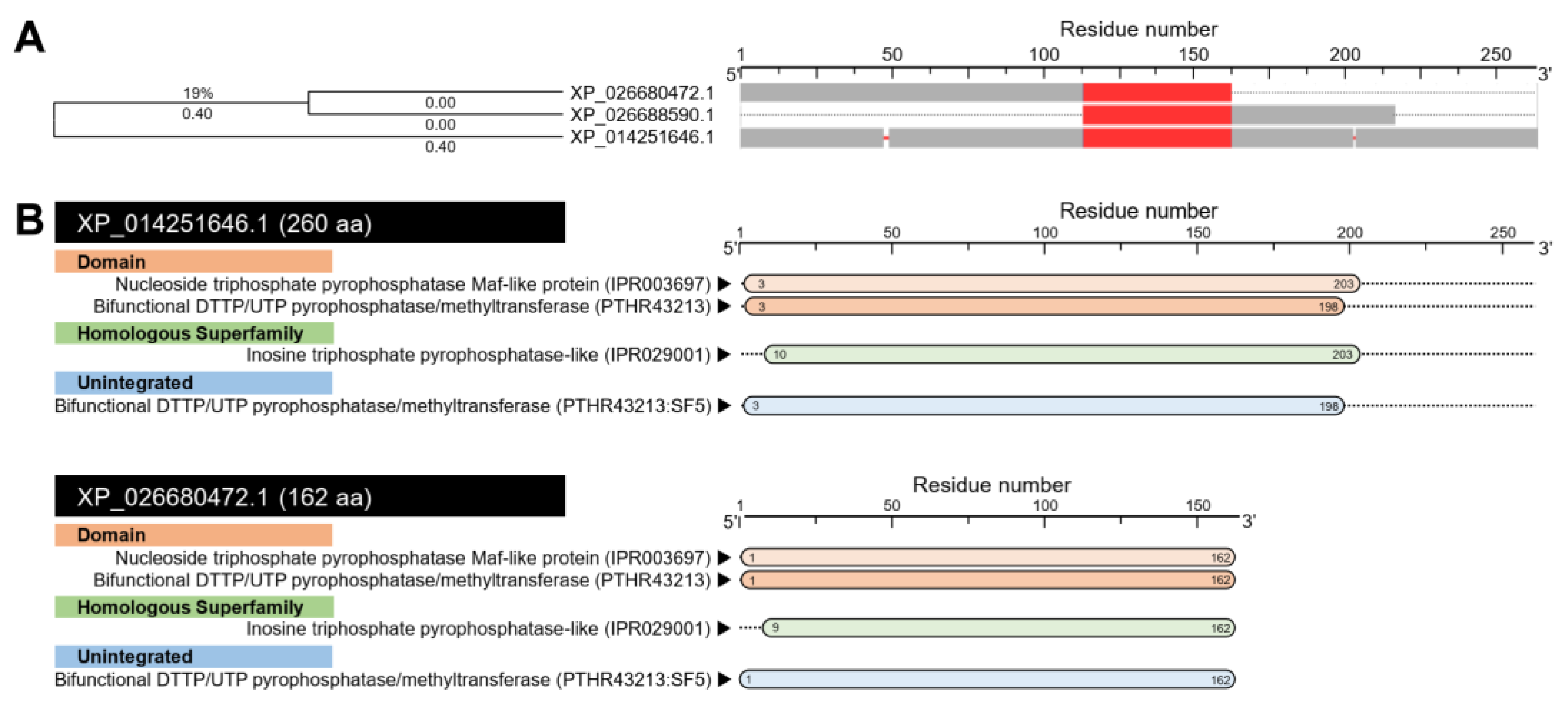
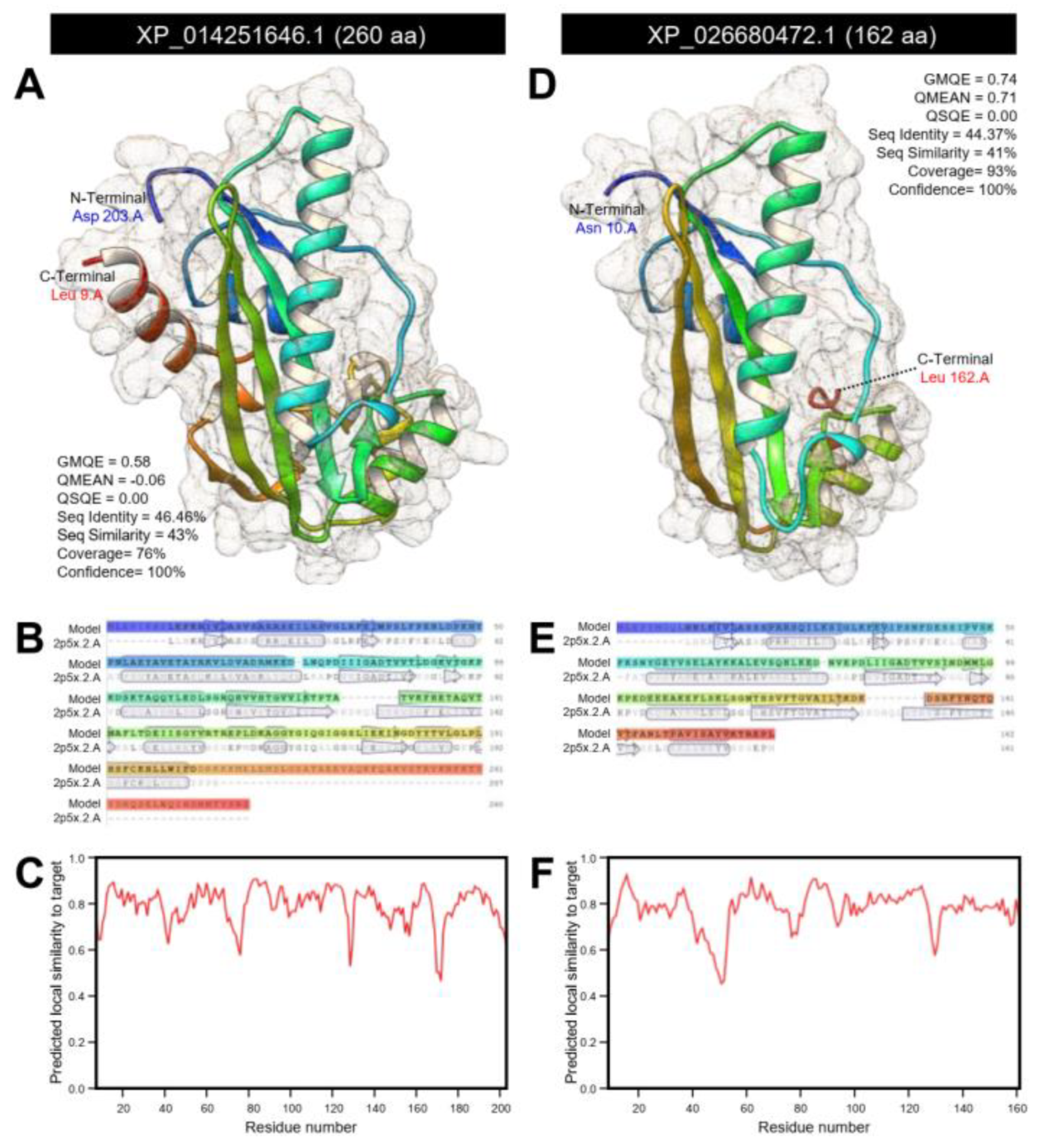
3.1.4. D. citri Genome Encodes for a Putative N-acetylserotonin O-methyltransferase (DcASMT)
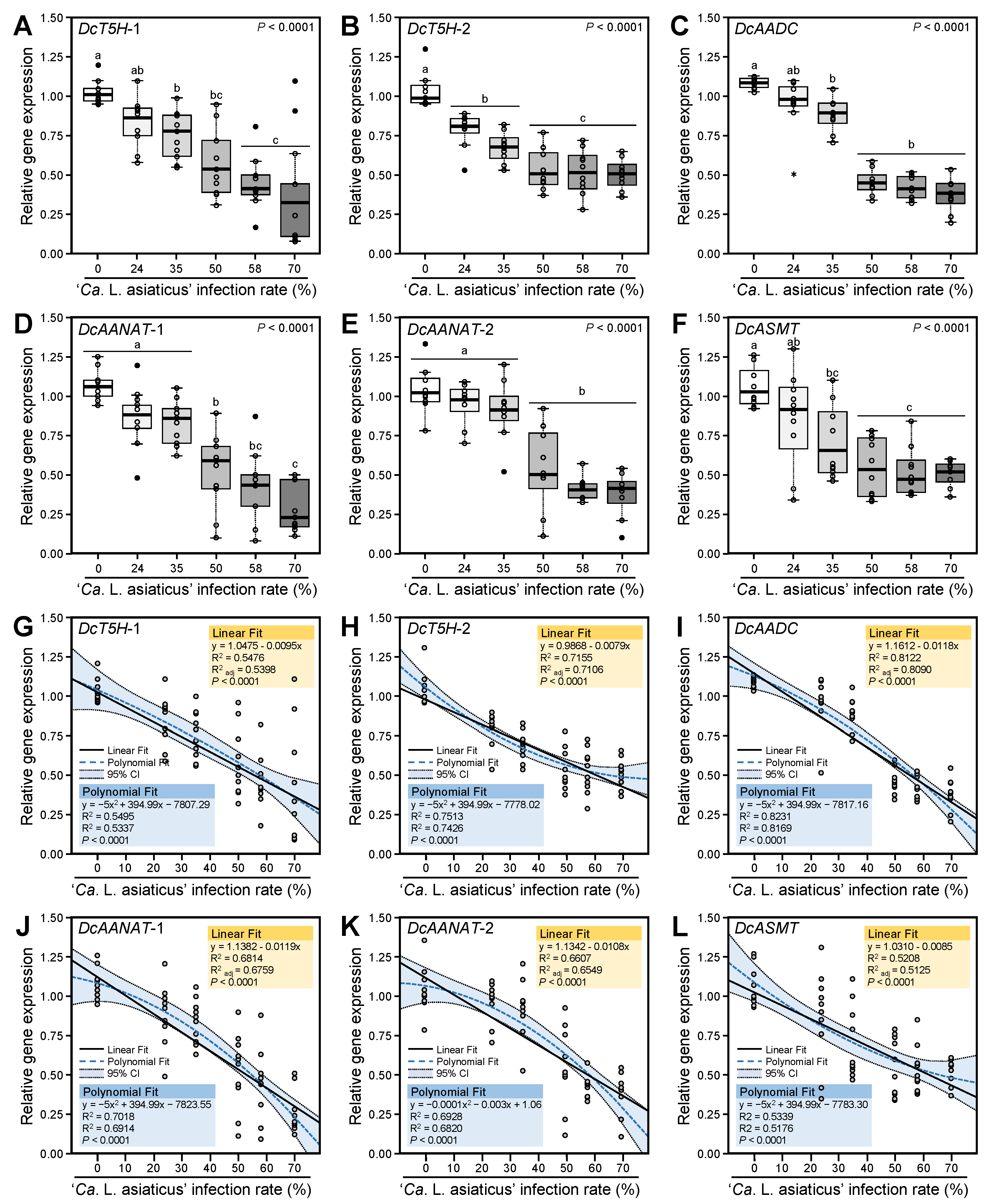
3.2. Ca. L. asiaticus Infection Downregulated the Expression of Melatonin Biosynthesis-Related Genes of D. citri
3.3. Expression Levels of Melatonin Biosynthesis-Related Genes of D. citri Are Negatively Correlated with the Ca. L. asiaticus Infection Rates
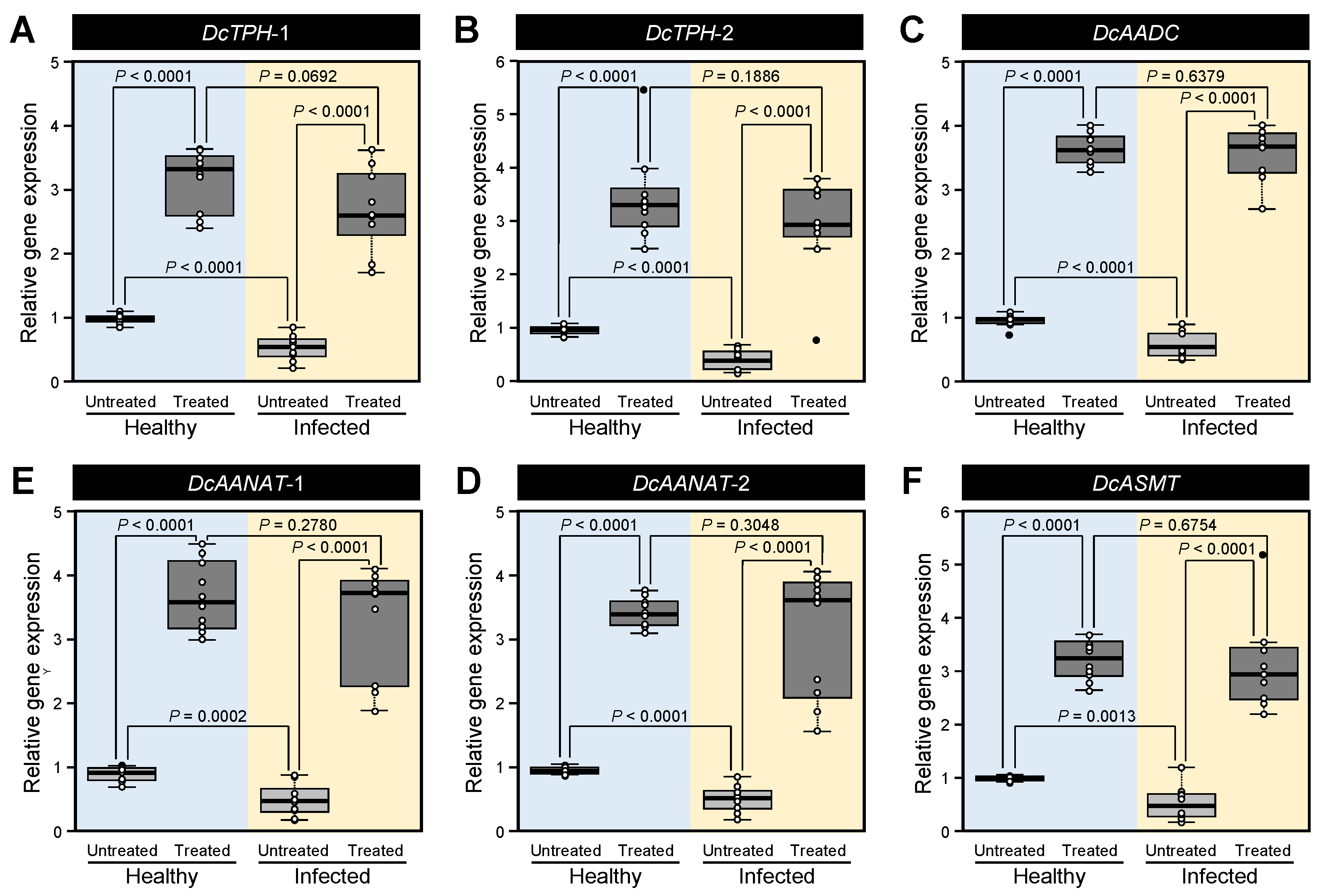
3.4. Melatonin Supplementation Induced the Expression Levels of Melatonin Biosynthesis-Related Genes of D. citri
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bové, J.M. Huanglongbing: A destructive, newly-emerging, century-old diesease of citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Gottwald, T.R. Current epidemiological understanding of citrus Huanglongbing. Annu. Rev. Phytopathol. 2010, 48, 119–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Trivedi, P. Citrus huanglongbing: A newly relevant disease presents unprecedented challenges. Phytopathology 2013, 103, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehela, Y.; Killiny, N. Revisiting the complex pathosystem of huanglongbing: Deciphering the role of citrus metabolites in symptom development. Metabolites 2020, 10, 409. [Google Scholar] [CrossRef]
- Singerman, A.; Rogers, M.E. The economic challenges of dealing with citrus greening: The case of Florida. J. Integr. Pest Manag. 2020, 11, pmz037. [Google Scholar] [CrossRef] [Green Version]
- Grafton-Cardwell, E.E.; Stelinski, L.L.; Stansly, P.A. Biology and management of Asian citrus psyllid, vector of the huanglongbing pathogens. Annu. Rev. Entomol. 2013, 58, 413–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosavljevic, I.; Schall, K.; Hoddle, C.; Morgan, D.; Hoddle, M. Biocontrol program targets Asian citrus psyllid in California’s urban areas. Calif. Agric. 2017, 71, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Milne, A.E.; Gottwald, T.; Parnell, S.R.; Alonso Chavez, V.; van den Bosch, F. What makes or breaks a campaign to stop an invading plant pathogen? PLOS Comput. Biol. 2020, 16, e1007570. [Google Scholar] [CrossRef] [Green Version]
- Halbert, S.E.; Manjunath, K.L. Asian citrus psyllids (Sternorrhyncha: Psyllidae) and greening disease of citrus: A literature review and assessment of risk in Florida. Florida Entomol. 2004, 87, 330–353. [Google Scholar] [CrossRef]
- Pelz-Stelinski, K.S.; Killiny, N. Better together: Association with ‘Candidatus liberibacter asiaticus’ increases the reproductive fitness of its insect vector, Diaphorina citri (Hemiptera: Liviidae). Ann. Entomol. Soc. Am. 2016, 48, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.; Pelz-Stelinski, K.; Stelinski, L.L. Effect of Candidatus Liberibacter asiaticus infection on susceptibility of Asian citrus psyllid, Diaphorina citri, to selected insecticides. Pest Manag. Sci. 2011, 67, 94–99. [Google Scholar] [CrossRef]
- Martini, X.; Hoffmann, M.; Coy, M.R.; Stelinski, L.L.; Pelz-Stelinski, K.S. Infection of an insect vector with a bacterial plant pathogen increases its propensity for dispersal. PLoS ONE 2015, 10, e0129373. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Killiny, N. Huanglongbing pathogen Candidatus Liberibacter asiaticus exploits the energy metabolism and host defence responses of its vector Diaphorina citri. Physiol. Entomol. 2017, 42, 319–335. [Google Scholar] [CrossRef]
- Killiny, N.; Nehela, Y.; Hijaz, F.; Vincent, C.I. A plant pathogenic bacterium exploits the tricarboxylic acid cycle metabolic pathway of its insect vector. Virulence 2018, 9, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Nehela, Y.; Killiny, N. Infection with phytopathogenic bacterium inhibits melatonin biosynthesis, decreases longevity of its vector, and suppresses the free radical-defense. J. Pineal Res. 2018, 65, e12511. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. Melatonin: The chemical expression of darkness. Mol. Cell. Endocrinol. 1991, 79, C153–C158. [Google Scholar] [CrossRef]
- Ikegami, K.; Yoshimura, T. Circadian clocks and the measurement of daylength in seasonal reproduction. Mol. Cell. Endocrinol. 2012, 349, 76–81. [Google Scholar] [CrossRef]
- Nehela, Y.; Killiny, N. Melatonin is involved in citrus response to the pathogen huanglongbing via modulation of phytohormonal biosynthesis. Plant Physiol. 2020, 184, 2216–2239. [Google Scholar] [CrossRef] [PubMed]
- Cipolla-Neto, J.; Amaral, F.G.; Afeche, S.C.; Tan, D.X.; Reiter, R.J. Melatonin, energy metabolism, and obesity: A review. J. Pineal Res. 2014, 56, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Vico, A.; Lardone, P.J.; Alvarez-Sánchez, N.; Rodríguez-Rodríguez, A.; Guerrero, J.M. Melatonin: Buffering the immune system. Int. J. Mol. Sci. 2013, 14, 8638–8683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Liang, F.; Zheng, J.; Zhou, K.; Chen, S.; Yu, J.; Zhang, J. Melatonin regulates apoptosis and autophagy via ROS-MST1 pathway in subarachnoid hemorrhage. Front. Mol. Neurosci. 2018, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Potes, Y.; Shabeeb, D.; Musa, A.E. Modulation of apoptosis by melatonin for improving cancer treatment efficiency: An updated review. Life Sci. 2019, 228, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Wu, P.; Seely, D.; Guyatt, G. Melatonin in the treatment of cancer: A systematic review of randomized controlled trials and meta-analysis. J. Pineal Res. 2005, 39, 360–366. [Google Scholar] [CrossRef]
- Zhao, M.; Wan, J.; Zeng, K.; Tong, M.; Lee, A.C.; Ding, J.; Chen, Q. The reduction in circulating melatonin level may contribute to the pathogenesis of ovarian cancer: A retrospective study. J. Cancer 2016, 7, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.P.; Lamarque, A.L.; Comba, A.; Berra, M.A.; Silva, R.A.; Labuckas, D.O.; Das, U.N.; Eynard, A.R.; Pasqualini, M.E. Synergistic anti-tumor effects of melatonin and PUFAs from walnuts in a murine mammary adenocarcinoma model. Nutrition 2015, 31, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin synthesis and function: Evolutionary history in animals and plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Back, K.; Tan, D.-X.; Reiter, R.J. Melatonin biosynthesis in plants: Multiple pathways catalyze tryptophan to melatonin in the cytoplasm or chloroplasts. J. Pineal Res. 2016, 61, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Byeon, Y.; Choi, G.-H.; Lee, H.Y.; Back, K. Melatonin biosynthesis requires N-acetylserotonin methyltransferase activity of caffeic acid O-methyltransferase in rice. J. Exp. Bot. 2015, 66, 6917–6925. [Google Scholar] [CrossRef] [Green Version]
- Byeon, Y.; Lee, H.J.; Lee, H.Y.; Back, K. Cloning and functional characterization of the Arabidopsis N-acetylserotonin O-methyltransferase responsible for melatonin synthesis. J. Pineal Res. 2016, 60, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lee, K.; Park, S.; Byeon, Y.; Back, K. Molecular cloning of rice serotonin N-acetyltransferase, the penultimate gene in plant melatonin biosynthesis. J. Pineal Res. 2013, 55, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Hardeland, R.; Back, K.; Manchester, L.C.; Alatorre-Jimenez, M.A.; Reiter, R.J. On the significance of an alternate pathway of melatonin synthesis via 5-methoxytryptamine: Comparisons across species. J. Pineal Res. 2016, 61, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Back, K. Overexpression of rice serotonin N-acetyltransferase 1 in transgenic rice plants confers resistance to cadmium and senescence and increases grain yield. J. Pineal Res. 2017, 62, e12392. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Byeon, Y.; Lee, K.; Lee, H.J.; Back, K. Cloning of Arabidopsis serotonin N-acetyltransferase and its role with caffeic acid O-methyltransferase in the biosynthesis of melatonin in vitro despite their different subcellular localizations. J. Pineal Res. 2014, 57, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Byeon, Y.; Lee, H.Y.; Back, K. Cloning and characterization of the serotonin N-acetyltransferase-2 gene (SNAT2) in rice (Oryza sativa). J. Pineal Res. 2016, 61, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.H.; Chen, F.; Zhu, B.Q.; Ma, L.Y.; Li, L.; Li, J.M. Molecular cloning and expression of gene encoding aromatic amino acid decarboxylase in “Vidal blanc” grape berries. Mol. Biol. Rep. 2012, 39, 4319–4325. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bian, L.; Jiao, Z.; Yu, K.; Wan, Y.; Zhang, G.; Guo, D. Molecular cloning and characterization of a grapevine (Vitis vinifera L.) serotonin N-acetyltransferase (VvSNAT2) gene involved in plant defense. BMC Genom. 2019, 20, 880. [Google Scholar] [CrossRef]
- Park, S.; Byeon, Y.; Lee, H.Y.; Kim, Y.-S.; Ahn, T.; Back, K. Cloning and characterization of a serotonin N-acetyltransferase from a gymnosperm, loblolly pine (Pinus taeda). J. Pineal Res. 2014, 57, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sumi-Ichinose, C.; Ichinose, H.; Nagatsu, T.; Takahashi, E.I.; Hon, T.A. Molecular cloning of genomic DNA and chromosomal assignment of the gene for human aromatic L-Amino acid decarboxylase, the enzyme for catecholamine and serotonin biosynthesis. Biochemistry 1992, 31, 2229–2238. [Google Scholar] [CrossRef]
- Kowlessur, D.; Kaufman, S. Cloning and expression of recombinant human pineal tryptophan hydroxylase in Escherichia coli: Purification and characterization of the cloned enzyme. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999, 1434, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Wessel, T.C.; Stone, D.M.; Carver, C.H.; Joh, T.H.; Park, D.H. Molecular cloning and characterization of cDNA encoding tryptophan hydroxylase from rat central serotonergic neurons. Mol. Brain Res. 1991, 9, 277–283. [Google Scholar]
- Taketoshi, M.; Horio, Y.; Imamura, I.; Tanaka, T.; Fukui, H.; Wada, H. Molecular cloning of guinea-pig aromatic-L-amino acid decarboxylase cDNA. Biochem. Biophys. Res. Commun. 1990, 170, 1229–1235. [Google Scholar] [CrossRef]
- Florez, J.C.; Seidenman, K.J.; Barrett, R.K.; Sangoram, A.M.; Takahashi, J.S. Molecular cloning of chick pineal tryptophan hydroxylase and circadian oscillation of its mRNA levels. Mol. Brain Res. 1996, 42, 25–30. [Google Scholar] [CrossRef]
- Bellipanni, G.; Rink, E.; Bally-Cuif, L. Cloning of two tryptophan hydroxylase genes expressed in the diencephalon of the developing zebrafish brain. Mech. Dev. 2002, 119, S215–S220. [Google Scholar] [CrossRef]
- Rahman, M.S.; Thomas, P. Molecular cloning, characterization and expression of two tryptophan hydroxylase (TPH-1 and TPH-2) genes in the hypothalamus of Atlantic croaker: Down-regulation after chronic exposure to hypoxia. Neuroscience 2009, 158, 751–765. [Google Scholar] [CrossRef]
- Ma, T.; Tao, J.; Yang, M.; He, C.; Tian, X.; Zhang, X.; Zhang, J.; Deng, S.; Feng, J.; Zhang, Z.; et al. An AANAT/ASMT transgenic animal model constructed with CRISPR/Cas9 system serving as the mammary gland bioreactor to produce melatonin-enriched milk in sheep. J. Pineal Res. 2017, 63, e12406. [Google Scholar] [CrossRef]
- Neckameyer, W.S.; White, K. A single locus encodes both phenylalanine hydroxylase and tryptophan hydroxylase activities in Drosophila. J. Biol. Chem. 1992, 267, 4199–4206. [Google Scholar] [CrossRef]
- Coleman, C.M.; Neckameyer, W.S. Serotonin synthesis by two distinct enzymes in Drosophila melanogaster. Arch. Insect Biochem. Physiol. 2005, 59, 12–31. [Google Scholar] [CrossRef]
- Hahn, S.L.; Hahn, M.; Joh, T.H. Genomic organization of the rat aromatic L-amino acid decarboxylase (AADC) locus: Partial analysis reveals divergence from the Drosophila dopa decarboxylase (DDC) gene structure. Mamm. Genome 1991, 1, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Davidson, N. Isolation and characterization of the dopa decarboxylase gene of Drosophila melanogaster. Mol. Cell. Biol. 1981, 1, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatarenkov, A.; Ayala, F.J. Nucleotide variation at the dopa decarboxylase (Ddc) gene in natural populations of Drosophila melanogaster. J. Genet. 2007, 86, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, E.; Jenö, P.; Meyer, U.A. Isolation and characterization of an arylalkylamine N -acetyltransferase from Drosophila melanogaster. FEBS Lett. 1995, 375, 148–150. [Google Scholar] [CrossRef] [Green Version]
- Hintermann, E.; Grieder, N.C.; Amherd, R.; Brodbeck, D.; Meyer, U.A. Cloning of an arylalkylamine N-acetyltransferase (aaNAT1) from Drosophila melanogaster expressed in the nervous system and the gut. Proc. Natl. Acad. Sci. USA 1996, 93, 12315–12320. [Google Scholar] [CrossRef] [Green Version]
- Amherd, R.; Hintermann, E.; Walz, D.; Affolter, M.; Meyer, U.A. Purification, cloning, and characterization of a second arylalkylamine N-Acetyltransferase from Drosophila melanogaster. DNA Cell Biol. 2000, 19, 697–705. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schaffer, A.A.; Yu, Y.-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Flores-Gonzalez, M.; Hosmani, P.S.; Fernandez-Pozo, N.; Mann, M.; Humann, J.L.; Main, D.; Heck, M.; Brown, S.; Mueller, L.A.; Saha, S. Citrusgreening.org: An open access and integrated systems biology portal for the Huanglongbing (HLB) disease complex. bioRxiv 2019, 868364. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, D.R.; Jeffries, K.A.; Bond, J.D.; Carpenter, A.M.; Rodriguez-Ospina, S.; Breydo, L.; Caswell, K.K.; Merkler, D.J. Mechanistic and structural analysis of Drosophila melanogaster arylalkylamine N-acetyltransferases. Biochemistry 2014, 53, 7777–7793. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Bjellqvist, B.; Basse, B.; Olsen, E.; Celis, J.E. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis 1994, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Tatineni, S.; Sagaram, U.S.; Gowda, S.; Robertson, C.J.; Dawson, W.O.; Iwanami, T.; Wang, N. In planta distribution of “Candidatus Liberibacter asiaticus” as revealed by polymerase chain reaction (PCR) and real-time PCR. Phytopathology 2008, 98, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.J.; KrishnaRaj, S.; Saxena, P.K. Tryptophan is a precursor for melatonin and serotonin biosynthesis in in vitro regenerated St. John’s wort (Hypericum perforatum L. cv. Anthos) plants. Plant Cell Rep. 2000, 19, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete genome sequence of citrus huanglongbing bacterium, “Candidatus Liberibacter asiaticus” obtained through metagenomics. Mol. Plant. Microbe Interact. 2009, 22, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Erlandsen, H.; Haavik, J.; Knappskog, P.M.; Stevens, R.C. Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry 2002, 41, 12569–12574. [Google Scholar] [CrossRef] [PubMed]
- Grenett, H.E.; Ledley, F.D.; Reed, L.L.; Woo, S.L. Full-length cDNA for rabbit tryptophan hydroxylase: Functional domains and evolution of aromatic amino acid hydroxylases. Proc. Natl. Acad. Sci. USA 1987, 84, 5530–5534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, L.; Byck, S.; Prevost, L.; Scriver, C.R. PAH Mutation Analysis Consortium Database: A database for disease-producing and other allelic variation at the human PAH locus. Nucleic Acids Res. 1996, 24, 125–126. [Google Scholar] [CrossRef] [Green Version]
- Erlandsen, H.; Hough, E.; Fusetti, F.; Stevens, R.C.; Fusetti, F.; Stevens, R.C.; Martinez, A.; Flatmark, T.; Fusetti, F. Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria. Nat. Struct. Biol. 1997, 4, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.R.C.; Tavares, G.D.B.; de Freitas, M.C.; De Mesquita, J.F. In silico analysis of the tryptophan hydroxylase 2 (TPH2) protein variants related to psychiatric disorders. PLoS ONE 2020, 15, e0229730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwill, K.E.; Sabatier, C.; Marks, C.; Raag, R.; Fitzpatrick, P.F.; Stevens, R.C. Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited neurodegenerative diseases. Nat. Struct. Biol. 1997, 4, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Sandmeier, E.; Hale, T.I.; Christen, P. Multiple evolutionary origin of pyridoxal-5′-phosphate-dependent amino acid decarboxylases. Eur. J. Biochem. 1994, 221, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Mizuguchi, H.; Nishino, J.; Hayashi, H.; Kagamiyama, H. Functionally important residues of aromatic L-amino acid decarboxylase probed by sequence alignment and site-directed mutagenesis. J. Biochem. 1996, 120, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Jackson, F.R. Prokaryotic and eukaryotic pyridoxal-dependent decarboxylases are homologous. J. Mol. Evol. 1990, 31, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Joseph, D.R.; Sullivan, P.M.; Wang, Y.M.; Kozak, C.; Fenstermacher, D.A.; Behrendsen, M.E.; Zahnow, C.A. Characterization and expression of the complementary DNA encoding rat histidine decarboxylase. Proc. Natl. Acad. Sci. USA 1990, 87, 733–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, L.; Galluccio, M.; Scalise, M.; Parravicini, C.; Palazzolo, L.; Eberini, I.; Indiveri, C. Novel insights into the transport mechanism of the human amino acid transporter LAT1 (SLC7A5). Probing critical residues for substrate translocation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodbeck, D.; Amherd, R.; Callaerts, P.; Hintermann, E.; Meyer, U.A.; Affolter, M. Molecular and biochemical characterization of the aaNAT1 (Dat) locus in Drosophila melanogaster: Differential expression of two gene products. DNA Cell Biol. 1998, 17, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.C. Arylalkylamine N-acetyltransferase: “The timezyme”. J. Biol. Chem. 2007, 282, 4233–4237. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, J.; Weissbach, H. Purification and properties of hydroxyindole-O-methyl transferase. J. Biol. Chem. 1961, 236, 211–213. [Google Scholar] [CrossRef]
- Liu, T.; Borjigin, J. N-acetyltransferase is not the rate-limiting enzyme of melatonin synthesis at night. J. Pineal Res. 2005, 39, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Database a | D. citri Genome Database b | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Description | Gene ID | mRNA | Protein | Protein–Protein Alignment Statistics | Gene Description | mRNA | Protein | Protein–Protein Alignment Statistics | ||||||||||||
| Accession | bp | Accession | aa | Theoretical Isoelectric Point (pI) | Molecular Weight (MW) | Max Score | Total Score | Query Cover (%) | E Value | Identity (%) | Accession | bp | Accession | aa | Identities (%) | Positives (%) | E Value | |||
| DcT5Hc | DcT5H | |||||||||||||||||||
| Tryptophan 5-hydroxylase 1-like | LOC113470334 | XM_026828703.1 | 1343 | XP_026684504.1 | 379 | 5.64 | 43,291.92 | 464 | 464 | 51 | 1 × 10−161 | 75.69 | Tryptophan 5-hydroxylase, putative | DcitrC076520.1.1 | 591 | DcitrP076520.1.1 | 196 | 100 | 100 | 7 × 10−143 |
| Protein henna-like | LOC103524631 | XM_017449691.2 | 1298 | XP_017305180.1 | 319 | 5.67 | 36,344.09 | 363 | 363 | 58 | 5 × 10−123 | 56.17 | Protein henna | DcitrC012845.1.1 | 1884 | DcitrP012845.1.1 | 627 | 87.15 | 87 | 0.0 |
| DcAADCd | DcAADC (also known as DcDDC) | |||||||||||||||||||
| Aromatic-L-amino-acid decarboxylase isoform X1 | LOC103520978 | XM_008486080.3 | 1939 | XP_008484302.1 | 481 | 5.70 | 54,429.72 | 734 | 734 | 90 | 0.0 | 75.27 | Dopa decarboxylase | DcitrC031955.1.1 | 1446 | DcitrP031955.1.1 | 481 | 100 | 100 | 0.0 |
| Aromatic-L-amino-acid decarboxylase | LOC103510318 | XM_017444526.2 | 1737 | XP_017300015.1 | 484 | 6.61 | 54825.40 | 692 | 692 | 90 | 0.0 | 71.61 | Dopa decarboxylase | DcitrC031955.1.1 | 1446 | DcitrP031955.1.1 | 481 | 93.83 | 94 | 0.0 |
| DcAANAT (also known as DcSNAT)e | DcAANAT (also known as DcSNAT) | |||||||||||||||||||
| Dopamine N-acetyltransferase-like isoform X1 | LOC103507708 | XM_026822511.1 | 1221 | XP_026678312.1 | 217 | 6.26 | 24,749.18 | 67.4 | 67.4 | 85 | 1 × 10−13 | 30.19 | Dopamine N-acetyltransferase | DcitrC025630.1.1 | 654 | DcitrP025630.1.1 | 217 | 99.08 | 99 | 8 × 10−161 |
| Dopamine N-acetyltransferase-like | LOC103507696 | XM_017443457.2 | 1911 | XP_017298946.1 | 220 | 5.39 | 24,808.39 | 114 | 114 | 83 | 2 × 10−31 | 34.78 | Dopamine N-acetyltransferase | DcitrC085745.1.1 | 723 | DcitrP085745.1.1 | 240 | 33.01 | 51.46 | 4 × 10−30 |
| DcASMTf | DcASMT | |||||||||||||||||||
| Septum formation protein Maf-like | LOC113468045 | XM_026824671.1 | 746 | XP_026680472.1 | 162 | 5.26 | 18,064.58 | 153 | 153 | 68 | 3 × 10−47 | 50.00 | N-acetylserotonin O-methyltransferase-like | DcitrC032285.1.1 | 825 | DcitrP032285.1.1 | 274 | 98.77 | 100 | 5 × 10−115 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nehela, Y.; Killiny, N. Diaphorina citri Genome Possesses a Complete Melatonin Biosynthesis Pathway Differentially Expressed under the Influence of the Phytopathogenic Bacterium, Candidatus Liberibacter asiaticus. Insects 2021, 12, 317. https://doi.org/10.3390/insects12040317
Nehela Y, Killiny N. Diaphorina citri Genome Possesses a Complete Melatonin Biosynthesis Pathway Differentially Expressed under the Influence of the Phytopathogenic Bacterium, Candidatus Liberibacter asiaticus. Insects. 2021; 12(4):317. https://doi.org/10.3390/insects12040317
Chicago/Turabian StyleNehela, Yasser, and Nabil Killiny. 2021. "Diaphorina citri Genome Possesses a Complete Melatonin Biosynthesis Pathway Differentially Expressed under the Influence of the Phytopathogenic Bacterium, Candidatus Liberibacter asiaticus" Insects 12, no. 4: 317. https://doi.org/10.3390/insects12040317
APA StyleNehela, Y., & Killiny, N. (2021). Diaphorina citri Genome Possesses a Complete Melatonin Biosynthesis Pathway Differentially Expressed under the Influence of the Phytopathogenic Bacterium, Candidatus Liberibacter asiaticus. Insects, 12(4), 317. https://doi.org/10.3390/insects12040317