
Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae)



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Genomic DNA Extraction
2.2. Sequence Assembly, Annotation and Bioinformatic Analysis
2.3. Sequence Alignment and Phylogenetic Analysis
3. Results and Discussion
3.1. Mitogenome Structure and Nucleotide Composition
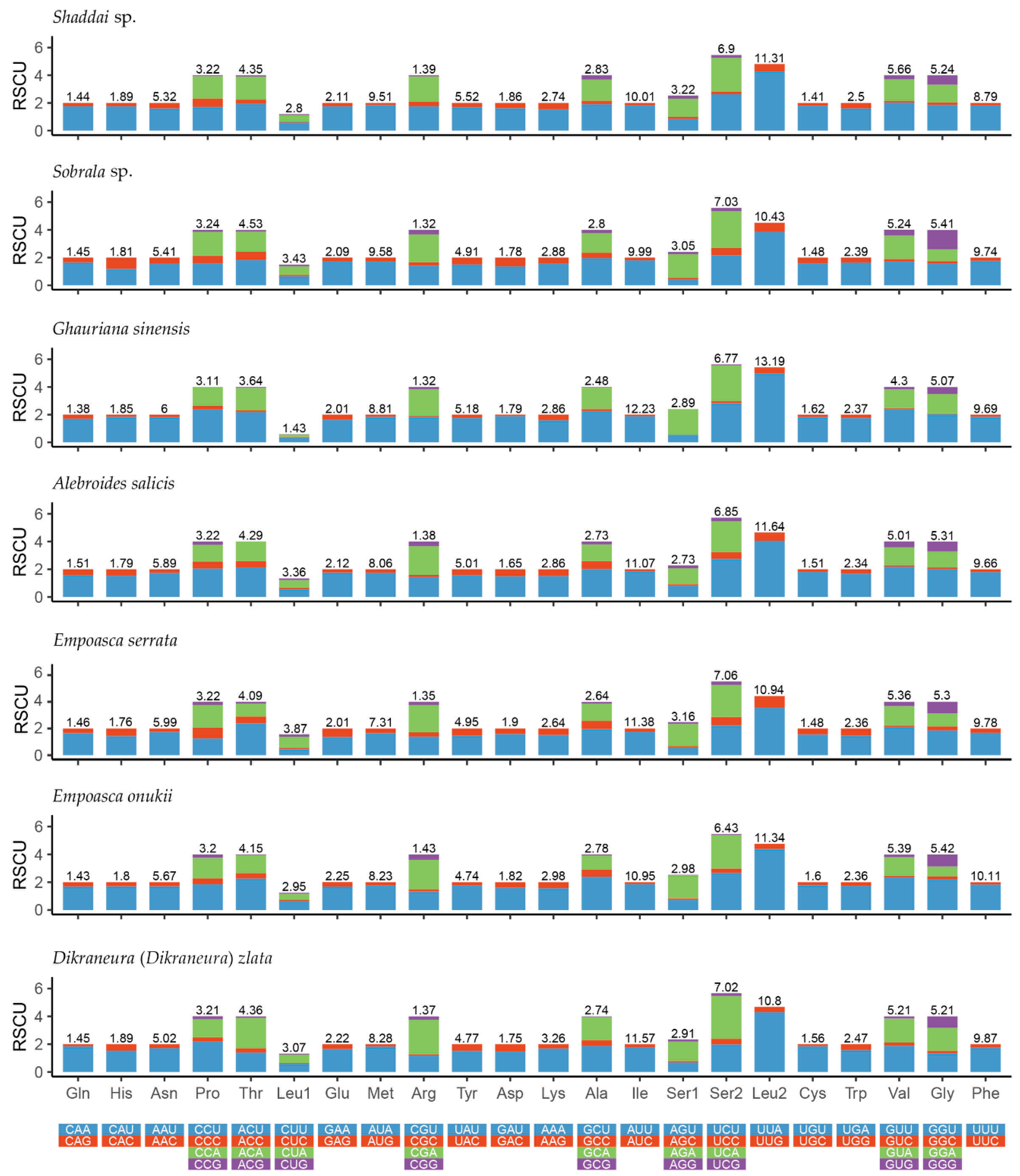
3.2. Protein-Coding Genes and Codon Usage
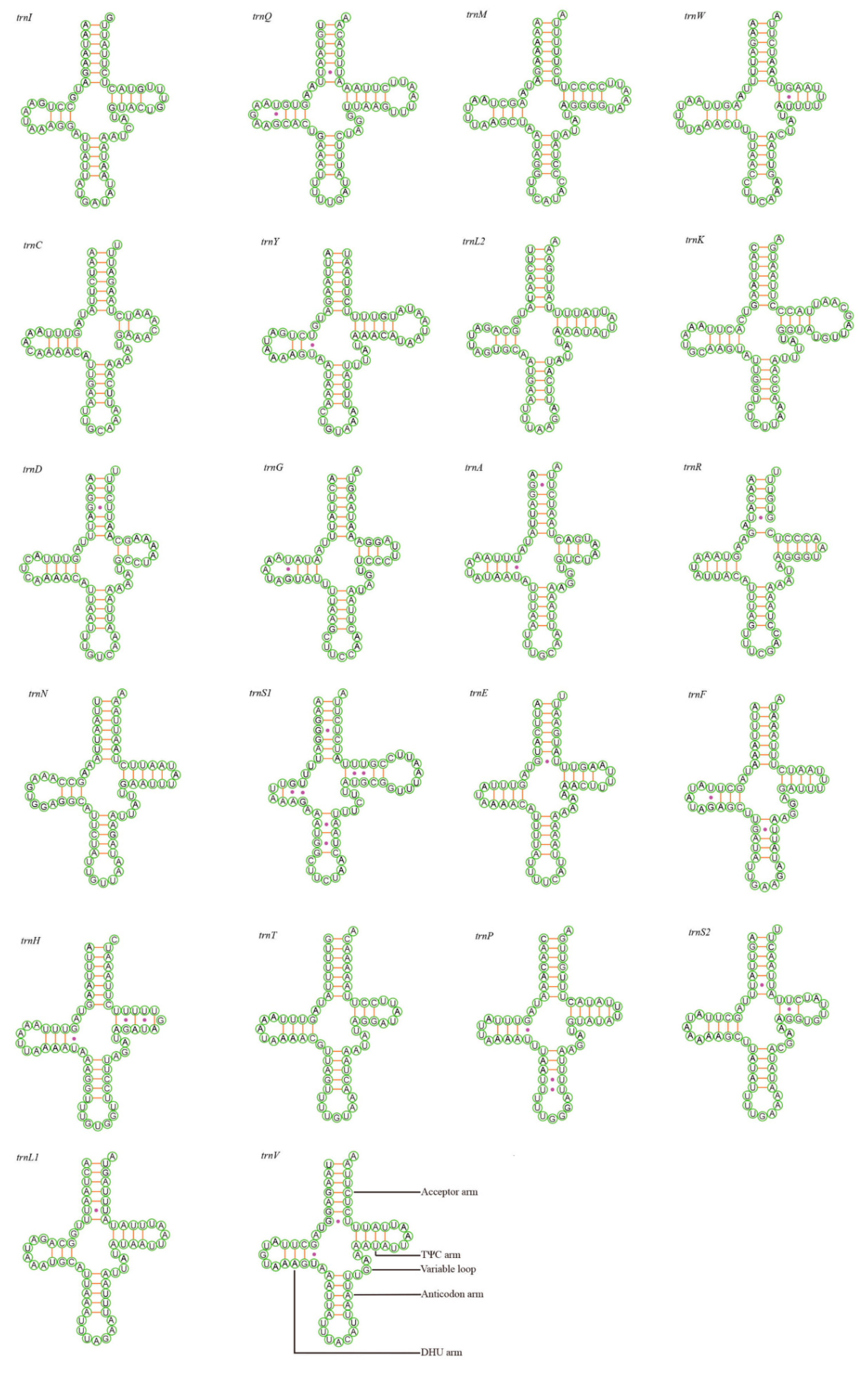
3.3. Transfer and Ribosomal RNA Genes
3.4. Control Region
3.5. Gene Overlaps and Intergenic Spacers
3.6. Nucleotide Diversity and Evolutionary Rate Analysis
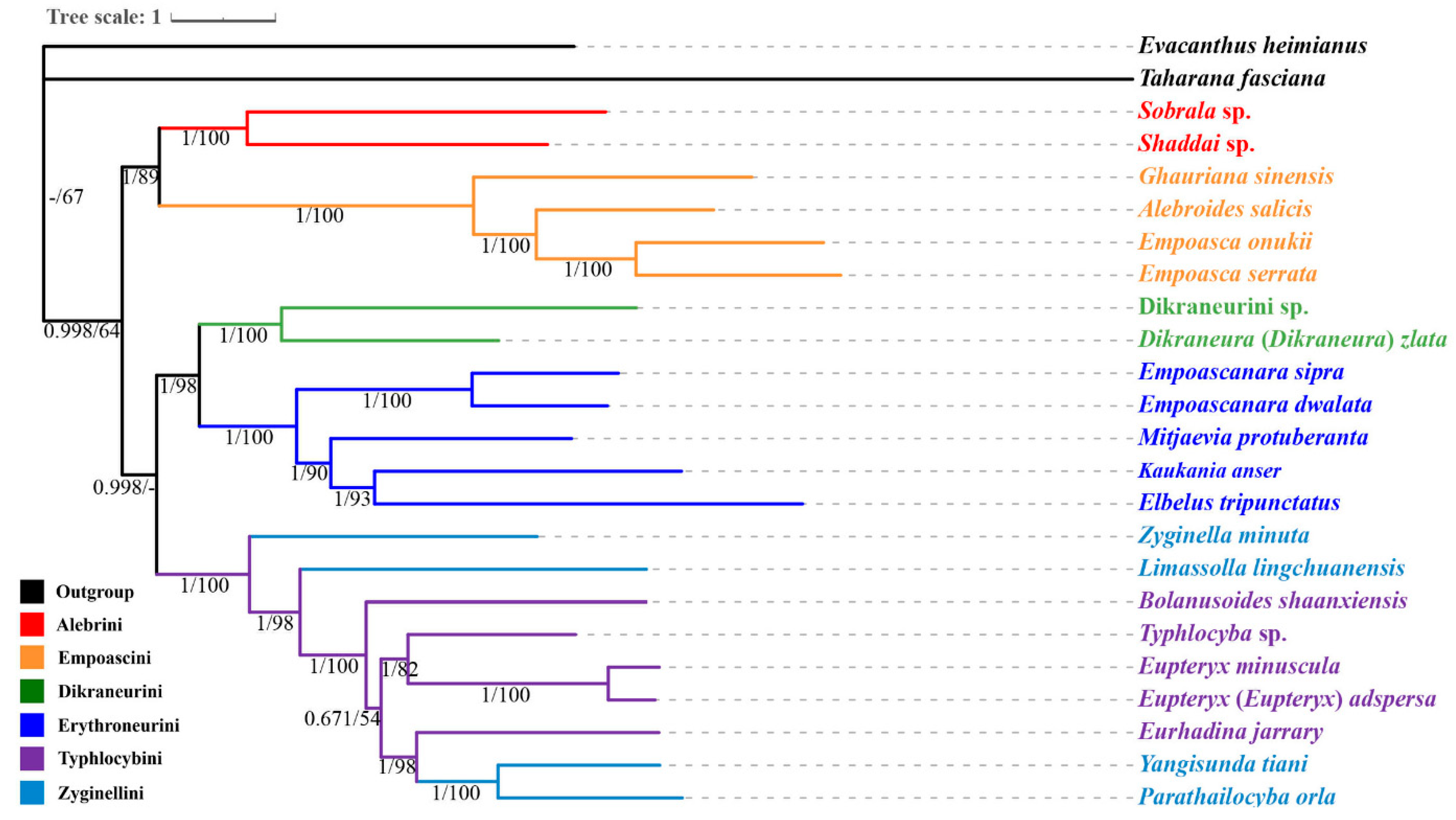
3.7. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dietrich, C.H.; Dmitriev, D.A. Review of the New World genera of the leafhopper tribe Erythroneurini (Hemiptera: Cicadellidae: Typhlocybinae). Bull. Ill. Nat. Hist. Surv. 2006, 37, 119–190. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Dmitriev, D.A. Review of the New World Erythroneurini (Hemiptera: Cicadellidae: Typhlocybinae). II. Genus Zyginama. Bull. Ill. Nat. Hist. Surv. 2008, 38, 129–175. [Google Scholar] [CrossRef]
- Dworakowska, I. The leafhopper tribe Zyginellini (Homoptera, Auchenorrhyncha, Cicadellidae, Typhlocybinae). Rev. Zool. Afr. 1979, 93, 299–331. [Google Scholar]
- Balme, G.R. In Phylogeny and Systematics of the Leafhopper Subfamily Typhlocybinae (Insecta: Hemiptera: Typhlocybinae). Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2007. [Google Scholar]
- Dietrich, C.H. Phylogeny of the leafhopper subfamily Evacanthinae with a review of Neotropical species and notes on related groups (Hemiptera: Membracoidea: Cicadellidae). Syst. Entomol. 2004, 29, 455–487. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Allen, J.M.; Lemmon, A.R.; Lemmon, E.M.; Takiya, D.M.; Evangelista, O.; Walden, K.K.; Grady, P.G.; Johnson, K.P.; Wiegmann, B. Anchored hybrid enrichment-based phylogenomics of leafhoppers and treehoppers (Hemiptera: Cicadomorpha: Membracoidea). Insect Syst. Divers. 2017, 1, 57–72. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Rakitov, R.A.; Holmes, J.L.; Black, W.C. Phylogeny of the major lineages of Membracoidea (Insecta: Hemiptera: Cicadomorpha) based on 28S rDNA Sequences. Mol. Phylogenet. Evol. 2001, 18, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.H. South American leafhoppers of the tribe Typhlocybini (Hemiptera: Cicadellidae: Typhlocybinae). Zoologia 2013, 30, 519–568. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L. A Taxonomic Study of Chinese Cicadellidae (Homoptera); Tianze Eldonejo: Yangling, China, 1990. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 1992, 2, 918–925. [Google Scholar] [CrossRef]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. Surv. Cell Biol. 1992, 141, 173–216. [Google Scholar]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, Y. Characterization of two complete mitochondrial genomes of Ledrinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Insects 2020, 11, 609. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Peng, X.X.; Jing, S.L.; Liu, B.F.; Zhu, L.L.; He, G.C. Intraspecific and interspecific variations in the mitochondrial genomes of Nilaparvata (Hemiptera: Delphacidae). J. Econ. Entomol. 2015, 108, 2021–2029. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.P.; Bu, C.P. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS ONE 2012, 7, e48778. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Liang, A. The complete mitochondrial genome sequence of Geisha distinctissima (Hemiptera: Flatidae) and comparison with other hemipteran insects. Acta Biochim. Biophys. Sin. 2009, 41, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Yu, T.; Zhang, Y. Characterization of the complete mitochondrial genome of Drabescus ineffffectus and Roxasellana stellata (Hemiptera: Cicadellidae: Deltocephalinae: Drabescini) and Their Phylogenetic Implications. Insects 2020, 11, 534. [Google Scholar] [CrossRef]
- Zhou, X.; Dietrich, C.H.; Huang, M. Characterization of the complete mitochondrial genomes of two species with preliminary investigation on phylogenetic status of Zyginellini (Hemiptera: Cicadellidae: Typhlocybinae). Insects 2020, 11, 684. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovli´c, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H. Complete mitochondrial genome of Evacanthus heimianus (Hemiptera: Cicadellidae: Evacanthinae) from China. Mitochondrial DNA Part B 2019, 4, 284–285. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.J.; Dai, R.H. Complete mitochondrial genome of Taharana fasciana (Insecta, Hemiptera: Cicadellidae) and comparison with other Cicadellidae insects. Genetica 2017, 145, 593–602. [Google Scholar] [CrossRef]
- Shi, R.; Yu, X.F.; Yang, M.F. Complete mitochondrial genome of Ghauriana sinensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 1367–1368. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.H.; Sun, C.Y.; Long, J.; Guo, J.J. Complete mitogenome of tea green leafhopper, Empoasca onukii (Hemiptera: Cicadellidae) from Anshun, Guizhou Province in China. Mitochondrial DNA Part B 2017, 2, 808–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.X.; Yuan, Z.W.; Li, C.; Song, Y.H. Complete mitochondrial genome sequence of Empoascanara dwalata (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 2260–2261. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Chen, X.X.; Li, C.; Song, Y.H. The complete mitochondrial genome of Empoascanara sipra (Hemiptera: Cicadellidae: Typhlocybinae) with phylogenetic consideration. Mitochondrial DNA Part B 2020, 5, 260–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Li, C.; Song, Y.H. Characterization of the complete mitochondrial genome of Mitjaevia protuberanta (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 601–602. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Yan, B.; Yu, X.F.; Yang, M.F. Complete mitochondrial genome of Zyginella minuta (Cicadellidae: Typhlocybinae: Zyginellini) from China, with its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 2795–2796. [Google Scholar] [CrossRef]
- Yuan, X.W.; Xiong, K.N.; Li, C.; Song, Y.H. The complete mitochondrial genome of Limassolla lingchuanensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 229–230. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Yuan, X.; Yuan, Z.; Song, Y.H. The complete mitochondrial genome of Parathailocyba orla (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 1981–1982. [Google Scholar] [CrossRef]
- Song, N.; Cai, W.Z.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 2017, 7, 10429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Haeseler, A.V.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.J.; Li, D.F.; Li, H.; Yang, M.F.; Dai, R.H. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci. Rep. 2019, 9, 15746. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Ding, X.L.; Zhang, Y.K.; Liu, J.; Chen, D.S.; Du, Y.; Hong, X.Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doublet, V.; Ubrig, E.; Alioua, A.; Bouchon, D.; Marcadé, I.; Maréchal-Drouard, L. Large gene overlaps and tRNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biol. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, Y.L.; Duan, Y.N. DNA barcoding of Deltocephalus Burmeister leafhoppers (Cicadellidae, Deltocephalinae, Deltocephalini) in China. Zookeys 2019, 867, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Zhang, Y.L. Two new species in the genus Destinoides (Hemiptera: Cicadellidae, Ledrinae) from China, with DNA barcoding data. Zootaxa 2019, 4711, 571–578. [Google Scholar] [CrossRef]
- Wang, Y.; Nansen, C.; Zhang, Y.L. Integrative insect taxonomy based on morphology, mitochondrial DNA, and hyperspectral reflectance profiling. Zool. J. Linn. Soc. 2016, 177, 378–394. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M. Biotaxonomy of typhlocybine leafhoppers of Pakistan. In Proceedings of the 1st International Workshop on Leafhoppers and Planthoppers (Auchenorrhyncha) of Economic Importance, London, UK, 4–7 October 1982; Commonwealth Institute of Entomology: London, UK, 1983; pp. 179–183. [Google Scholar]
- Chen, X.X.; Li, C.; Song, Y.H. The complete mitochondrial genomes of two erythroneurine leafhoppers (Hemiptera, Cicadellidae, Typhlocybinae, Erythroneurini) with assessment of the phylogenetic status and relationships of tribes of Typhlocybinae. Zookeys 2021, 1037, 137–159. [Google Scholar] [CrossRef]
- Young, D.A. A reclassification of Western Hemisphere Typhlocybinae (Homoptera, Cicadellidae). Univ. Kans. Sci. Bull. 1952, 35, 1–217. [Google Scholar] [CrossRef]
- Ruppel, R.F. A summary of the tribes proposed in Typhlocybinae (Hemiptera, Cicadellidae). Mich. Acad. 1987, 19, 29–35. [Google Scholar]
- Jiang, J.; Chen, X.X.; Li, C.; Song, Y.H. Mitogenome and phylogenetic analysis of typhlocybine leafoppers (Hemiptera: Cicadellidae). Sci. Rep. 2021, 11, 10053. [Google Scholar] [CrossRef]
- Xu, Y.; Dietrich, C.H.; Zhang, Y.L.; Dmitriev, D.A.; Zhang, L.; Wang, Y.M.; Lu, S.H.; Qin, D.Z. Phylogeny of the tribe Empoascini (Hemiptera: Cicadellidae: Typhlocybinae) based on morphological characteristics, with reclassifification of the Empoasca generic group. Syst. Entomol. 2021, 46, 266–286. [Google Scholar] [CrossRef]
- Anufriev, G.A. Les cicadellidaes de le Territoire Maritime. Horae Soc. Entomol. Unionis Sov. 1978, 60, 1–215. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Tribe | Species | Accession Number | Reference |
|---|---|---|---|---|---|
| Cicadellidae | Evacanthinae | Evacanthus heimianus | MG813486 | [31] | |
| Coelidiinae | Taharana fasciana | NC_036015 | [32] | ||
| Typhlocybinae | Alebrini | Shaddai sp. | MZ014457 | This study | |
| Sobrala sp. | MZ014458 | This study | |||
| Empoascini | Ghauriana sinensis | MN699874 | [33] | ||
| Alebroides salicis | MZ014449 | This study | |||
| Empoasca serrata | MZ014453 | This study | |||
| Empoasca onukii | NC_037210 | [34] | |||
| Dikraneurini | Dikraneura (Dikraneura) zlata | MZ014450 | This study | ||
| Dikraneurini sp. | MZ014451 | This study | |||
| Erythroneurini | Empoascanara dwalata | MT350235 | [35] | ||
| Empoascanara sipra | NC_048516 | [36] | |||
| Mitjaevia protuberanta | NC_047465 | [37] | |||
| Elbelus tripunctatus | MZ014452 | This study | |||
| Kaukania anser | MZ014456 | This study | |||
| Zyginellini | Zyginella minuta | MT488436 | [38] | ||
| Limassolla lingchuanensis | NC_046037 | [39] | |||
| Parathailocyba orla | MN894531 | [40] | |||
| Yangisunda tiani | MZ014459 | This study | |||
| Typhlocybini | Bolanusoides shaanxiensis | MN661136 | Unpublished | ||
| Typhlocyba sp. | KY039138 | [41] | |||
| Eupteryx (Eupteryx) adspersa | MZ014454 | This study | |||
| Eupteryx minuscula | MN910279 | Unpublished | |||
| Eurhadina jarrary | MZ014455 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Huang, M.; Zhang, Y. Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae). Insects 2021, 12, 678. https://doi.org/10.3390/insects12080678
Lin S, Huang M, Zhang Y. Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae). Insects. 2021; 12(8):678. https://doi.org/10.3390/insects12080678
Chicago/Turabian StyleLin, Shuanghu, Min Huang, and Yalin Zhang. 2021. "Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae)" Insects 12, no. 8: 678. https://doi.org/10.3390/insects12080678
APA StyleLin, S., Huang, M., & Zhang, Y. (2021). Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae). Insects, 12(8), 678. https://doi.org/10.3390/insects12080678