
Comparative Mitogenomic Analysis of Two Longhorn Beetles (Coleoptera: Cerambycidae: Lamiinae) with Preliminary Investigation into Phylogenetic Relationships of Tribes of Lamiinae


 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and DNA Extraction
2.2. Sequence Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
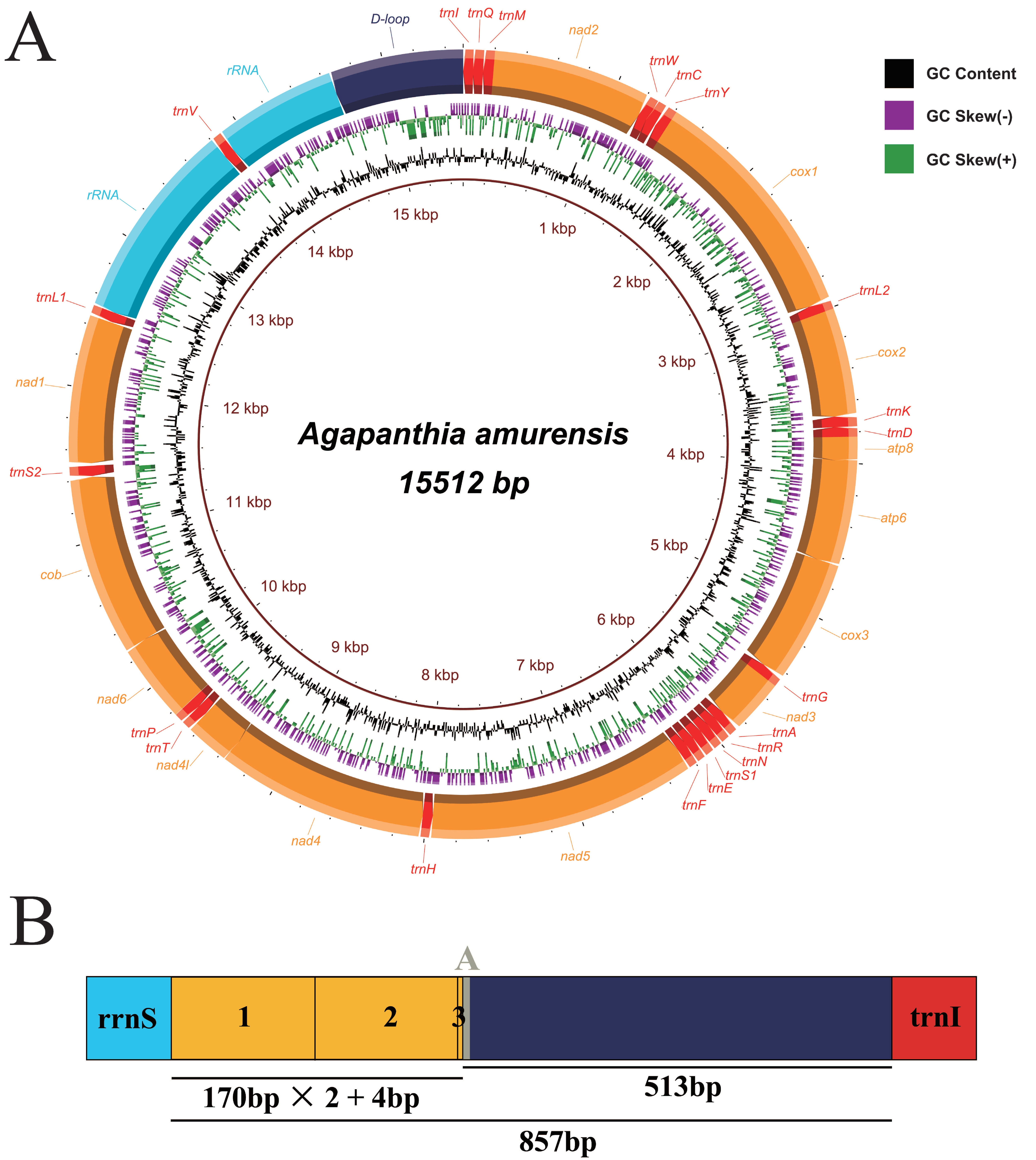
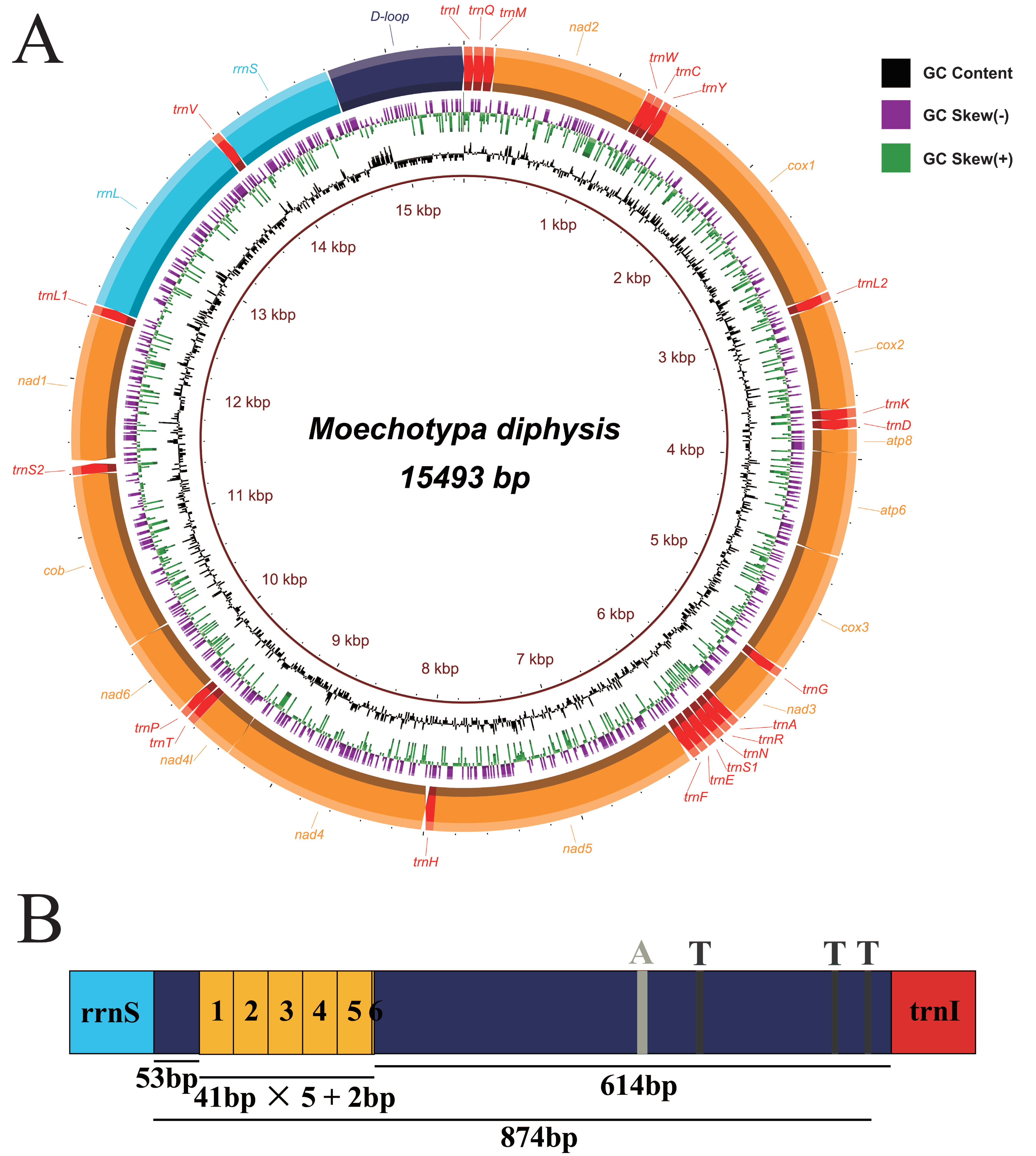
3.1. General Features of the Mitogenomes of Agapanthia amurensis and Moechotypa diphysis
3.2. Genome Structure
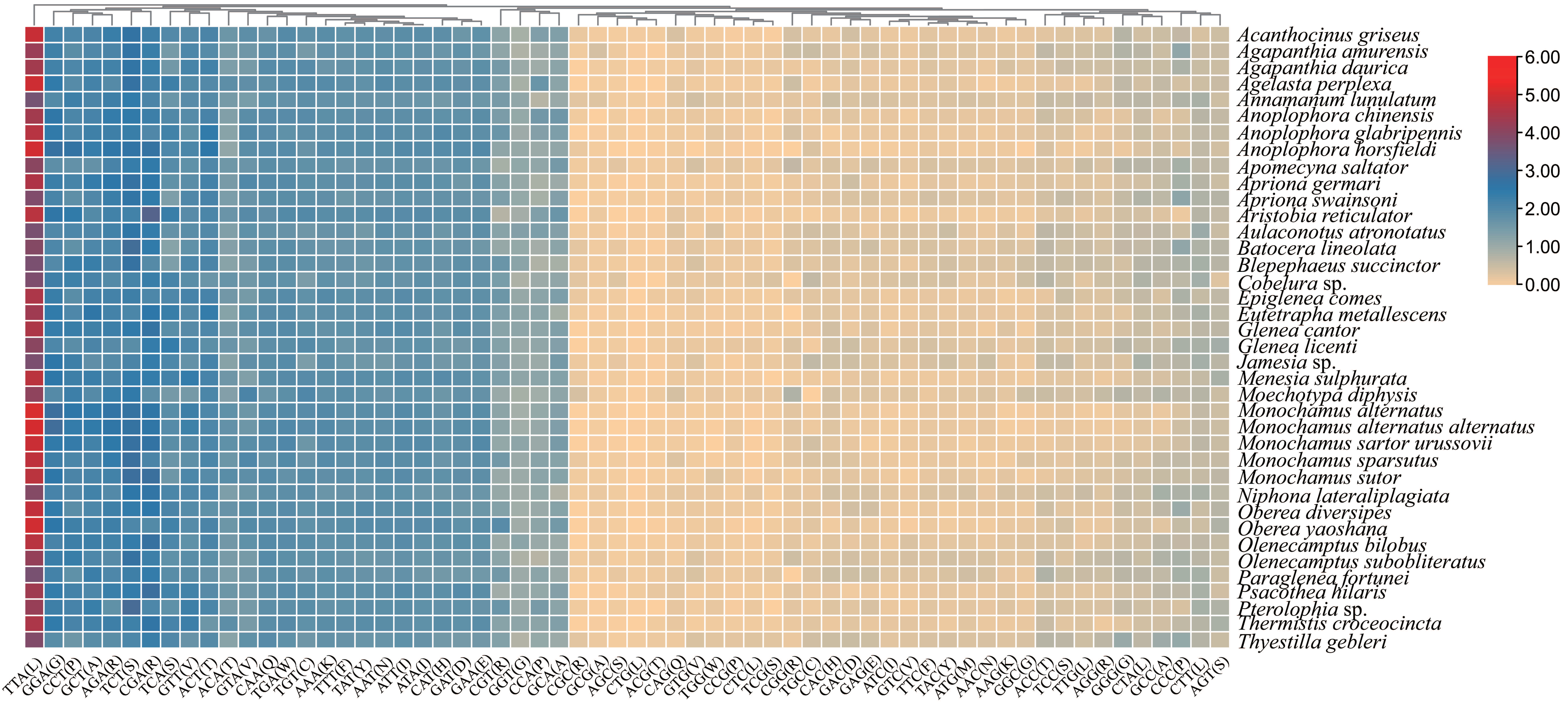
3.2.1. Protein-Coding Genes
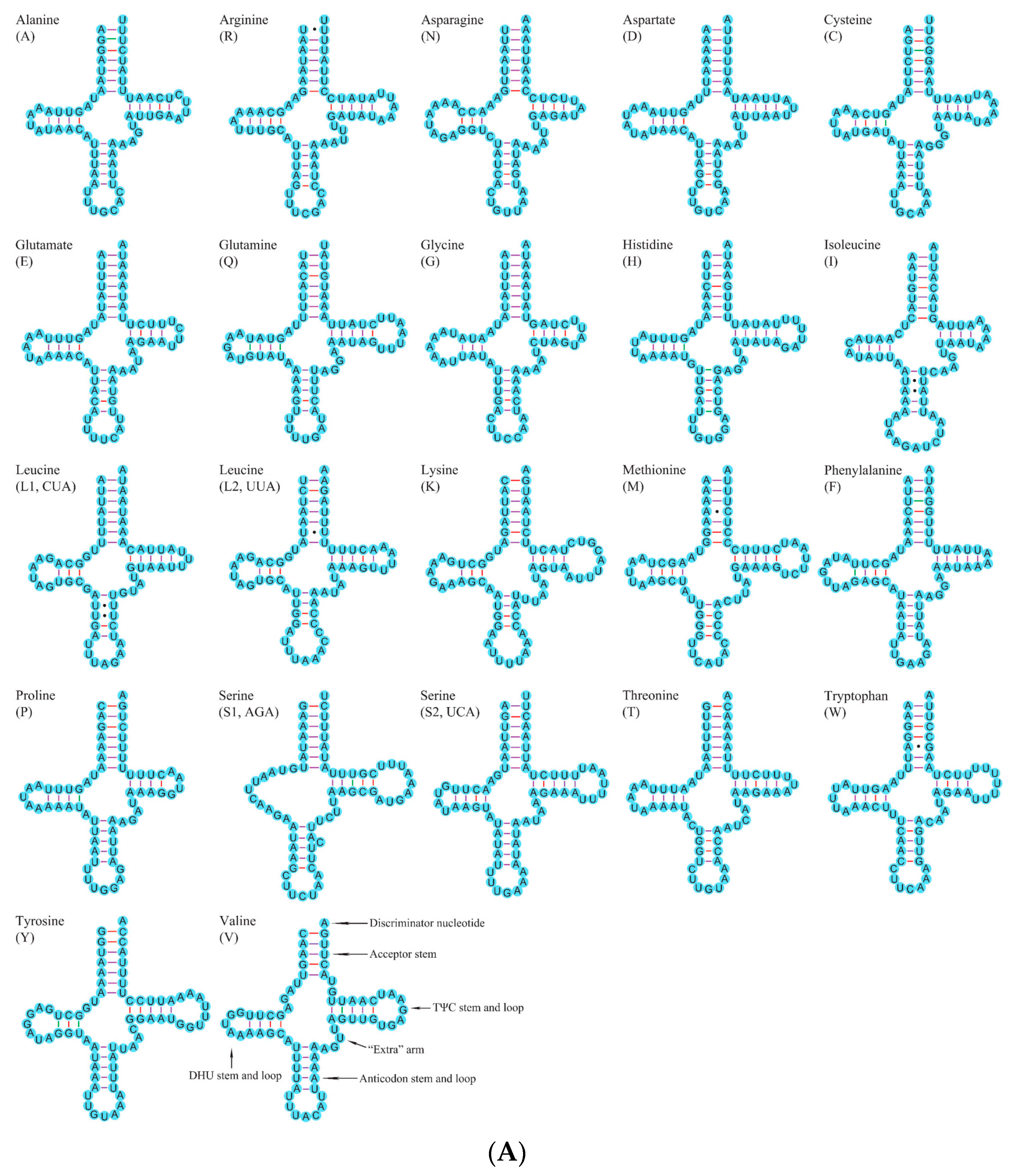
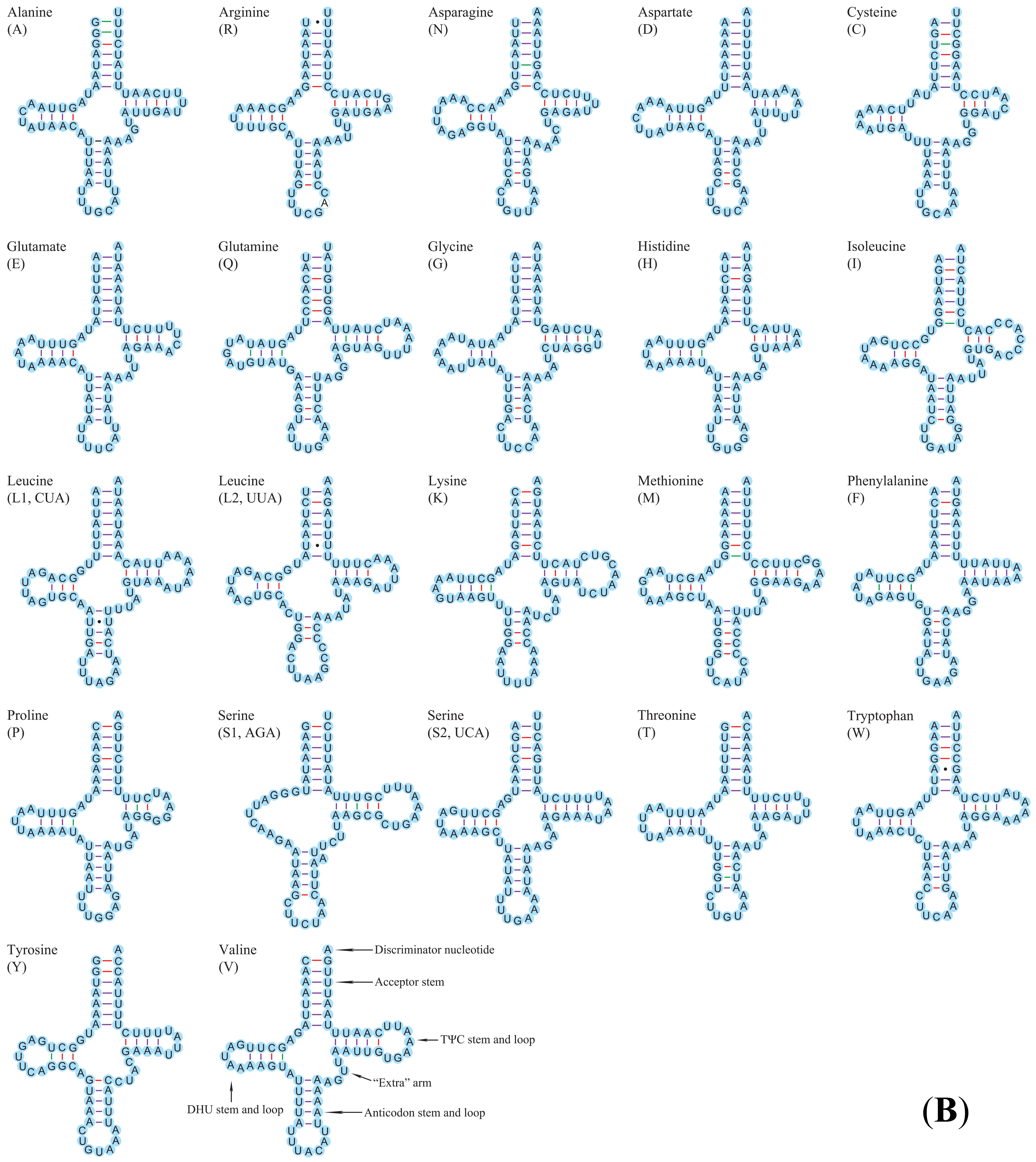
3.2.2. Transfer and Ribosomal RNA Genes
3.2.3. Control Region
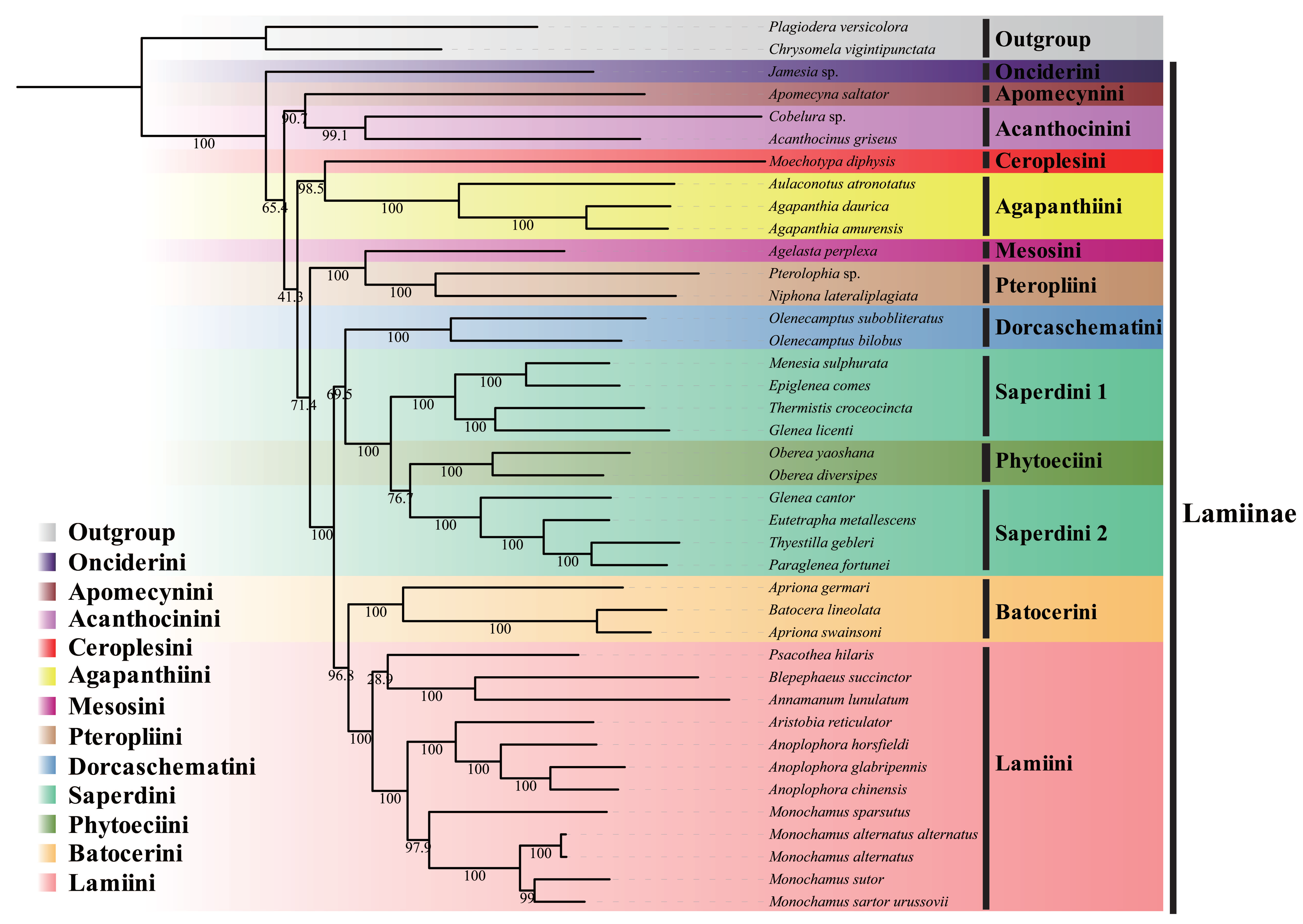
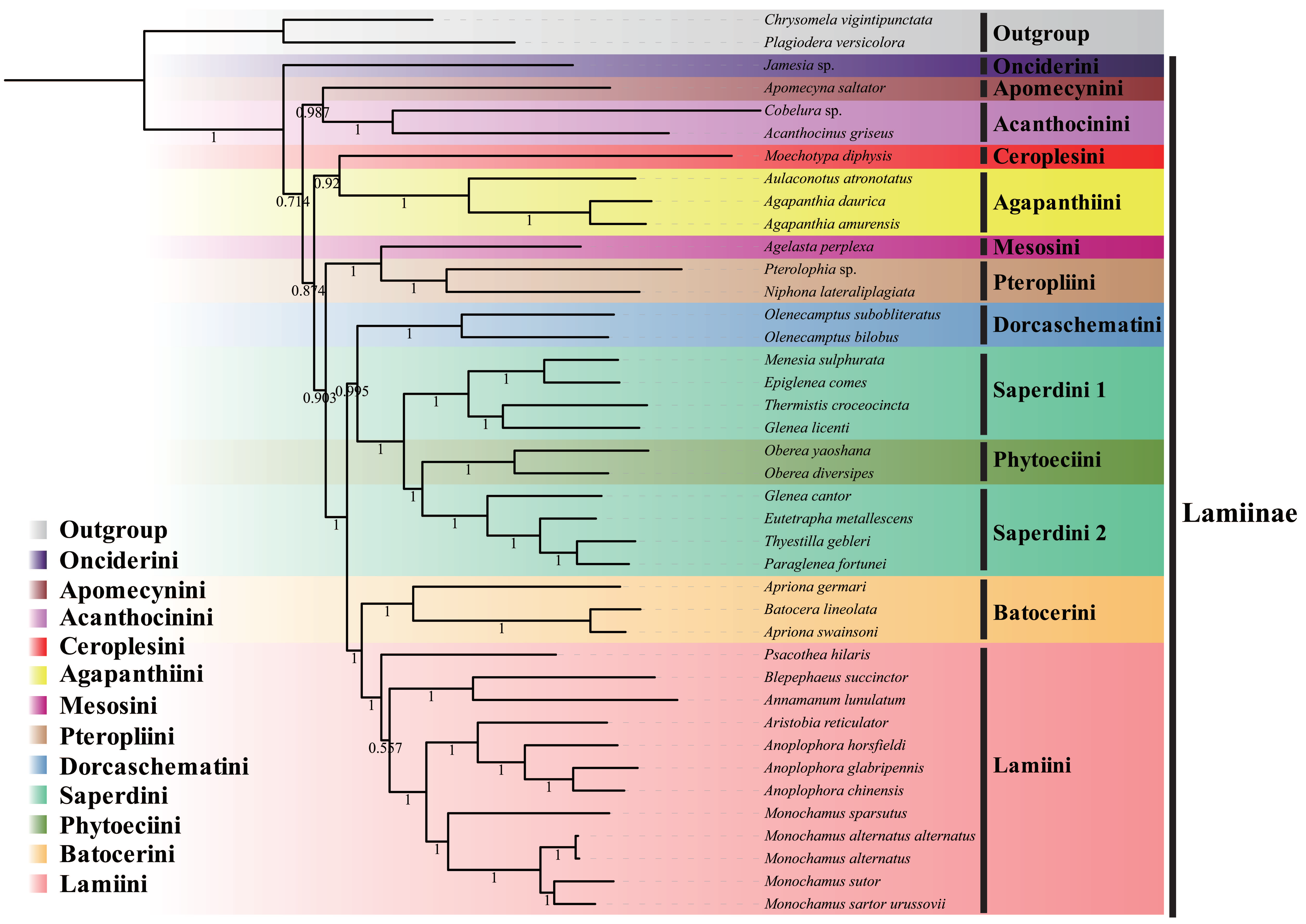
3.3. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tavakilian, G.L.; Chevillotte, H. Titan: Base de données internationales sur les Cerambycidae ou Longicornes. Version 3.0. Available online: http://titan.gbif.fr/index.html (accessed on 1 May 2021).
- Ikeda, T.; Oda, K. The occurrence of attractiveness for Monochamus alternatus Hope (Coleoptera: Cerambycidae) in nematode-infected pine trees. J. Jpn. For. Soc. 1980, 62, 432–434. [Google Scholar] [CrossRef]
- Evans, H.F.; McNamara, D.G.; Braasch, H.; Chadoeuf, J.; Magnusson, C. Pest risk analysis (PRA) for the territories of the European Union (as PRA area) on Bursaphelenchus xylophilus and its vectors in the genus Monochamus. EPPO Bull. 1996, 26, 199–249. [Google Scholar] [CrossRef]
- Jiang, S.N. Coleoptera, Fauna Sinica Insecta.Vol 21 Coleoptera Cerambycidae Lepturinae; Science Press: Beijing, China, 2001; pp. 1–299. ISBN 7-03-008163-3/Q934. [Google Scholar]
- Allison, J.D.; Borden, J.H.; Seybold, S.J. A review of the chemical ecology of the Cerambycidae (Coleoptera). Chemoecology 2004, 14, 123–150. [Google Scholar] [CrossRef]
- Raje, K.R.; Abdel-Moniem, H.E.M.; Farlee, L.; Ferris, V.B.; Holland, J.D. Abundance of pest and benign Cerambycidae both increase with decreasing forest productivity. Agric. For. Entomol. 2012, 14, 165–169. [Google Scholar] [CrossRef]
- Karpiński, L.; Maák, I.; Wegierek, P. The role of nature reserves in preserving saproxylic biodiversity: Using longhorn beetles (Coleoptera: Cerambycidae) as bioindicators. Eur. Zool. J. 2021, 88, 487–504. [Google Scholar] [CrossRef]
- Napp, D.S. Phylogenetic relationships among the subfamilies of Cerambycidae (Coleoptera, Chrysomeloidea). Rev. Bras. Entomol. 1994, 38, 265–419. [Google Scholar]
- Marvaldi, A.E.; Duckett, C.N.; Kjer, K.M.; Gillespie, J.J. Structural alignment of 18S and 28S rDNA sequences provides insights into the phylogeny of Phytophaga (Coleoptera: Curculionoidea and Chrysomeloidea). Zool. Scripta 2008, 38, 63–77. [Google Scholar] [CrossRef]
- Wang, B.; Ma, J.Y.; McKenna, D.D.; Yan, E.V.; Zhang, H.C.; Jarzembowski, E.A. The earliest known longhorn beetle (Cerambycidae: Prioninae) and implications for the early evolution of Chrysomeloidea. J. Syst. Palaeontol. 2013, 12, 565–574. [Google Scholar] [CrossRef]
- Haddad, S.; Shin, S.; Lemmon, A.R.; Lemmon, E.M.; Svacha, P.; Farrell, B.; Ślipiński, A.; Windsor, D.; Mckenna, D.D. Anchord hybrid enrichment provides new insights into the phylogeny and evolution of longhorned beetles (Cerambycidae). Syst. Entomol. 2017, 43, 68–89. [Google Scholar] [CrossRef]
- Nie, R.E.; Vogler, A.P.; Yang, X.K.; Lin, M.Y. Higher-level phylogeny of longhorn beetles (Coleoptera: Chrysomeloidea) inferred from mitochondrial genomes. Syst. Entomol. 2020, 46, 56–70. [Google Scholar] [CrossRef]
- Souza, D.D.S.; Marinoni, L.; Monné, M.L.; Gómez-Zurita, J. Molecular phylogenetic assessment of the tribal classification of Lamiinae (Coleoptera: Cerambycidae). Mol. Phylogenetics Evol. 2020, 145, 106736. [Google Scholar] [CrossRef]
- Latreille, P.A. Familles Naturelles du Règne Animal, Exposées Succinctement et Dans un Ordre Analytique, Avec l’Indication de Leurs Genres; Imprimerie DE J.: Paris, France, 1825; pp. 1–570. [Google Scholar]
- Blanchard, C.É. Histoire dês Insectes, Traitant de Leurs Moeurs et Leurs Métamorphoses em General et Comprenant une Nouvelle Classification Fondée sur Leurs Rapports Naturels; Hymenoptères et Coléoptères; Librairie de Firmin Didot Frères: Paris, France, 1845; pp. 154–161. [Google Scholar]
- Thomson, J. Essai d’Une Classification de la Famille des Cérambycides et Matériaux pour Servir a Une Monographie de Cette Famille; Bouchard-Huzard: Paris, France, 1860; pp. 1–128. [Google Scholar]
- Thomson, J. Systema Cerambycidarum ou Exposé de Tous les Genres Compris Dans La famille dês Cérambycides et Familles Limitrophes; Société royale des sciences de Liège: Paris, France, 1864; pp. 1–540. [Google Scholar]
- Aurivillius, C. Coleopterorum Catalogus, Pars 73, Cerambycidae: Lamiinae, I; W, Junk: Berlin, Germany, 1922; pp. 1–322. [Google Scholar]
- Aurivillius, C. Coleopterorum Catalogus, Pars 74, Cerambycidae: Lamiinae II; W, Junk: Berlin, Germany, 1923; pp. 322–704. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 1; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1958; pp. 1–48. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 2.; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1959; pp. 109–182. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 3.; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1960; pp. 183–284. [Google Scholar]
- Breuning, S. Révision des Pteropliini (Col. Cerambycidae). Pesquisas Zoologia 1961, 9, 5–60. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 4; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1961; pp. 287–382. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 5; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1961; pp. 387–459. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 6; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1962; pp. 463–555. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 7; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1963; pp. 49–107. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 8; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1965; pp. 559–655. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 9; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1966; pp. 659–765. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 10; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1967; pp. 771–864. [Google Scholar]
- Breuning, S. Catalogue des Lamiaires du Monde (Col. Céramb.). 11; Verlag des Museum, G. Frey: Tutzing bei Munich, Germany, 1969; pp. 865–1069. [Google Scholar]
- Bouchard, P.; Bousquet, Y.; Davies, A.; Alonso-Zarazaga, M.A.; Lawrence, J.F.; Lyal, C.H.C.; Newton, A.F.; Reid, C.A.M.; Schmitt, M.; Ślipiński, S.A.; et al. Family-group names in Coleoptera (Insecta). ZooKeys 2011, 88, 1–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccone, C.; Giorgi, C.D.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Abascal, F.; Posada, D.; Knight, R.D.; Zardoya, R. Parallel evolution of the genetic code in arthropod mitochondrial genomes. PLoS Biol. 2006, 4, e127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Gan, H.M.; Lee, Y.P.; Poore, G.C.; Austin, C.M. Digging deeper: New gene order rearrangements and distinct patterns of codons usage in mitochondrial genomes among shrimps from the Axiidea, Gebiidea and Caridea (Crustacea: Decapoda). PeerJ 2017, 5, e2982. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Li, R.M.; Song, X.; Du, Y.M. Mitochondrial genome of Aromia bungii (Coleoptera: Chrysomeloidea: Cerambycidae) and phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2012, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partition finder 2: New Methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, M.J.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Vogler, A.P. Family-level sampling of mitochondrial genomes in Coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2016, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, 242–245. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Chen, D.B.; Liu, H.H.; Hu, H.L.; Bian, H.X.; Zhang, R.S.; Yang, R.S.; Jiang, X.F.; Shi, S.L. The complete mitochondrial genome of the longhorn Beetle Dorysthenes paradoxus (Coleoptera: Cerambycidae: Prionini) and the implication for the phylogenetic relationships of the Cerambycidae species. J. Insect Sci. 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, X.Y.; Xu, X.D.; Zhang, Z.Y.; Zhang, J.Y. The complete mitochondrial genomes of five longicorn beetles (coleoptera: Cerambycidae) and phylogenetic relationships within cerambycidae. PeerJ 2019, 7, e7633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Èntomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Rand, D.M. Endotherms, ectotherms, and mitochondrial genome size variation. J. Mol. Evol. 1993, 37, 281–295. [Google Scholar] [CrossRef]
- Rydin, C.; Källersjö, M. Taxon sampling and seed plant phylogeny. Cladistics 2002, 18, 485–513. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Donoghue, M.J. Patterns of variation in levels of homoplasy. Evolution 1989, 43, 1781–1795. [Google Scholar] [CrossRef]
- Cai, C.; Tihelka, E.; Pisani, D.; Donoghue, P.C. Data curation and modeling of compositional heterogeneity in insect phylogenomics: A case study of the phylogeny of Dytiscoidea (Coleoptera: Adephaga). Mol. Phylogenetics Evol. 2020, 147, 106782. [Google Scholar] [CrossRef]
- Ai, D.; Peng, L.; Qin, D.; Zhang, Y. Characterization of three complete mitogenomes of Flatidae (Hemiptera: Fulgoroidea) and compositional heterogeneity analysis in the Planthoppers’ mitochondrial phylogenomics. Int. J. Mol. Sci. 2021, 22, 5586. [Google Scholar] [CrossRef]
- Nie, R.E.; Andújar, C.; Gómez-Rodríguez, C.; Bai, M.; Xue, H.J.; Tang, M.; Vogler, A.P. The phylogeny of leaf beetles (Chrysomelidae) inferred from mitochondrial genomes. Syst. Entomol. 2020, 45, 188–204. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Guan, J.Y.; Cao, Y.R.; Dai, X.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Mitogenome analysis of four lamiinae species (Coleoptera: Cerambycidae) and gene expression responses by monochamus alternatus when infected with the parasitic nematode, bursaphelenchus mucronatus. Insects 2021, 12, 453. [Google Scholar] [CrossRef] [PubMed]
- Cherepamov, A.I.; Zolotarenko, G.S.; Kothekar, V.S. Cerambycidae of Northern Asia. Vollume 3, Lamiinae, Part 1 (English Edition); Amerind Publishing: New Delhi, India, 1990; pp. 1–300. [Google Scholar]
- Löbl, I.; Smetana, A. Catalogue of Palaearctic Coleoptera, Vol. 6: Chrysomeloidae; Apollo Books: Stenstrup, Denmark, 2010; pp. 1–924. [Google Scholar]
- Gressitt, J.L. Longicorn Beetles of China. Longicornia, Études et Notes sur les Longicornes; Paul Lechevalier: Paris, Germany, 1951; pp. 1–667. Volume 2. [Google Scholar]
- Ohbayashi, N.; Niisato, T. Longicorn Beetles of Japan; Tokai University Press: Kanagawa, Japan, 2007; pp. 1–818. [Google Scholar]
- Toki, W.; Kubota, K. Molecular phylogeny based on mitochondrial genes and evolution of host plant use in the long-horned beetle tribe Lamiini (Coleoptera: Cerambycidae) in Japan. Environ. Entomol. 2010, 4, 1336–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; Lu, H.; Chen, L.; Sabatelli, S.; Wang, C.; Xie, G.; Wang, P.; Liu, M.; Wang, W.; Audisio, P. Comparative Mitogenomic Analysis of Two Longhorn Beetles (Coleoptera: Cerambycidae: Lamiinae) with Preliminary Investigation into Phylogenetic Relationships of Tribes of Lamiinae. Insects 2021, 12, 820. https://doi.org/10.3390/insects12090820
Ren Y, Lu H, Chen L, Sabatelli S, Wang C, Xie G, Wang P, Liu M, Wang W, Audisio P. Comparative Mitogenomic Analysis of Two Longhorn Beetles (Coleoptera: Cerambycidae: Lamiinae) with Preliminary Investigation into Phylogenetic Relationships of Tribes of Lamiinae. Insects. 2021; 12(9):820. https://doi.org/10.3390/insects12090820
Chicago/Turabian StyleRen, Yifang, Huanhuan Lu, Longyan Chen, Simone Sabatelli, Chaojie Wang, Guanglin Xie, Ping Wang, Meike Liu, Wenkai Wang, and Paolo Audisio. 2021. "Comparative Mitogenomic Analysis of Two Longhorn Beetles (Coleoptera: Cerambycidae: Lamiinae) with Preliminary Investigation into Phylogenetic Relationships of Tribes of Lamiinae" Insects 12, no. 9: 820. https://doi.org/10.3390/insects12090820
APA StyleRen, Y., Lu, H., Chen, L., Sabatelli, S., Wang, C., Xie, G., Wang, P., Liu, M., Wang, W., & Audisio, P. (2021). Comparative Mitogenomic Analysis of Two Longhorn Beetles (Coleoptera: Cerambycidae: Lamiinae) with Preliminary Investigation into Phylogenetic Relationships of Tribes of Lamiinae. Insects, 12(9), 820. https://doi.org/10.3390/insects12090820