
Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Data Retrieval from the Public Database
2.2. Constructing and Evaluating the Reference Transcriptome Sequence Data
2.3. Reference Transcriptome Sequence Annotations
2.4. Analysis of the Public RNA-Seq Data to Search for Novel Candidate Genes Involved in Immune Responses
3. Results
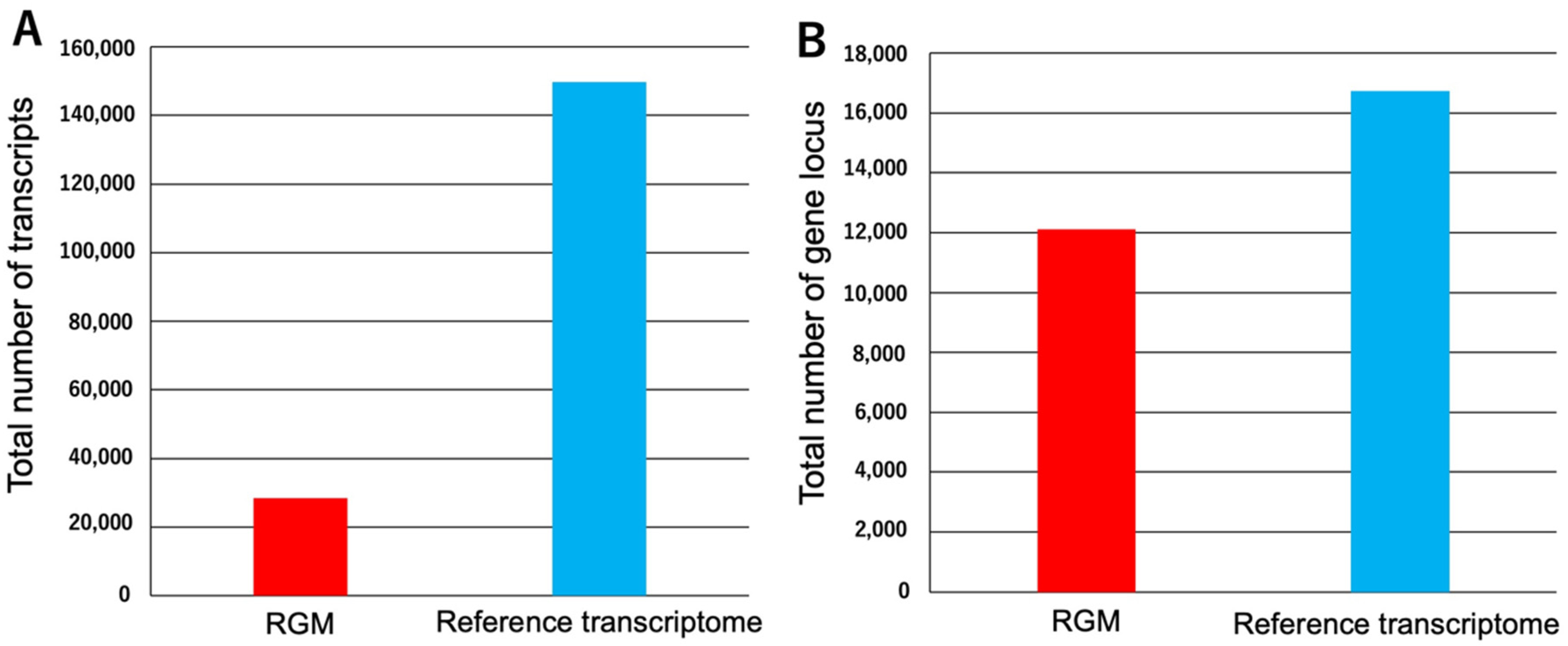
3.1. Construction of Reference Transcriptome of a Mellifera Using Public RNA-Seq Data
3.2. Meta-Analysis of Reference Transcriptome and Published RNA-Seq Data to Detect new Candidate Genes (Transcripts) Involved in the Immune Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winston, M. The Biology of the Honey Bee; Harvard University Press: Cambridge, MA, USA, 1991. [Google Scholar]
- Weinstock, G.M.; Robinson, G.E.; Gibbs, R.A.; Weinstock, G.M.; Weinstock, G.M.; Robinson, G.E.; Worley, K.C.; Evans, J.D.; Maleszka, R.; Robertson, H.M. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef] [Green Version]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüns, H.; Crozier, R.H. Relish regulates expression of antimicrobial peptide genes in the honeybee, Apis mellifera, shown by RNA interference. Insect Mol. Biol. 2007, 16, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Bono, H. All of gene expression (AOE): An integrated index for public gene expression databases. PLoS ONE 2020, 15, e0227076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fent, K.; Schmid, M.; Hettich, T.; Schmid, S. The neonicotinoid Thiacloprid causes transcriptional alteration of genes associated with mitochondria at environmental concentrations in honey bees. Environ. Pollut. 2020, 266, 115297. [Google Scholar] [CrossRef]
- Decio, P.; Ustaoglu, P.; Derecka, K.; Hardy, I.C.W.; Roat, T.C.; Malaspina, O.; Mongan, N.; Stöger, R.; Soller, M. Thiamethoxam exposure deregulates short ORF gene expression in the honey bee and compromises immune response to bacteria. Sci. Rep. 2021, 11, 1489. [Google Scholar] [CrossRef]
- Soares, M.P.M.; Pinheiro, D.G.; de Paula Freitas, F.C.; Simões, Z.L.P.; Bitondi, M.M.G. Transcriptome dynamics during metamorphosis of imaginal discs into wings and thoracic dorsum in Apis mellifera castes. BMC Genom. 2021, 22, 756. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, Y.B.; Barron, A.B.; Zeng, Z.J. Transcriptomic, morphological, and developmental comparison of adult honey bee queens (Apis mellifera) reared from eggs or worker larvae of differing ages. J. Econ. Entomol. 2020, 113, 2581–2587. [Google Scholar] [CrossRef]
- Kawamoto, M.; Jouraku, A.; Toyoda, A.; Yokoi, K.; Minakuchi, Y.; Katsuma, S.; Fujiyama, A.; Kiuchi, T.; Yamamoto, K.; Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem. Mol. Biol. 2019, 107, 53–62. [Google Scholar] [CrossRef]
- Yokoi, K.; Tsubota, T.; Jouraku, A.; Sezutsu, H.; Bono, H. Reference transcriptome data in silkworm Bombyx mori. Insects 2021, 12, 519. [Google Scholar] [CrossRef]
- Li, Q.; Li, M.; Zhu, M.; Zhong, J.; Wen, L.; Zhang, J.; Zhang, R.; Gao, Q.; Yu, X.Q.; Lu, Y. Genome-wide identification and comparative analysis of cry toxin receptor families in 7 insect species with a focus on Spodoptera litura. Insect Sci. 2022, 29, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, Y.; Cao, W.; Jouraku, A.; Sakai, H.; Sezutsu, H.; Yokoi, K. Co-expression network and time-course expression analyses to identify silk protein regulatory factors in Bombyx mori. Insects 2022, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Bono, H. Multi-Omic meta-analysis of transcriptomes and the Bibliome uncovers novel hypoxia-inducible genes. Biomedicines 2021, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ono, Y.; Bono, H. Comparison of oxidative and hypoxic stress responsive genes from meta-analysis of public transcriptomes. Biomedicines 2021, 9, 1830. [Google Scholar] [CrossRef]
- Yokoi, K.; Kimura, K.; Bono, H. Revealing Landscapes of Transposable Elements in Apis species by Meta-analysis. Insects 2022, 13, 698. [Google Scholar] [CrossRef]
- Doublet, V.; Poeschl, Y.; Gogol-Döring, A.; Alaux, C.; Annoscia, D.; Aurori, C.; Barribeau, S.M.; Bedoya-Reina, O.C.; Brown, M.J.F.; Bull, J.C.; et al. Unity in Defence: Honeybee Workers Exhibit Conserved Molecular Responses to Diverse Pathogens. BMC Genom. 2017, 18, 207. [Google Scholar] [CrossRef] [Green Version]
- Bono, H.; Sakamoto, T.; Kasukawa, T.; Tabunoki, H. Systematic functional annotation workflow for insects. Insects 2022, 13, 586. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Cornman, R.S.; Lopez, D.; Evans, J.D. Transcriptional response of honey bee larvae infected with the bacterial pathogen Paenibacillus larvae. PLoS ONE 2013, 8, e65424. [Google Scholar] [CrossRef] [Green Version]
- Ryabov, E.V.; Fannon, J.M.; Moore, J.D.; Wood, G.R.; Evans, D.J. The Iflaviruses Sacbrood virus and deformed wing virus evoke different transcriptional responses in the honeybee which may facilitate their horizontal or vertical transmission. PeerJ 2016, 4, e1591. [Google Scholar] [CrossRef] [Green Version]
- Azzouz-Olden, F.; Hunt, A.; DeGrandi-Hoffman, G. Transcriptional response of honey bee (Apis mellifera) to differential nutritional status and nosema infection. BMC Genom. 2018, 19, 628. [Google Scholar] [CrossRef]
- Rutter, L.; Carrillo-Tripp, J.; Bonning, B.C.; Cook, D.; Toth, A.L.; Dolezal, A.G. Transcriptomic responses to diet quality and viral infection in Apis mellifera. BMC Genom. 2019, 20, 412. [Google Scholar] [CrossRef]
- Chang, Z.T.; Huang, Y.F.; Chen, Y.W.; Yen, M.R.; Hsu, P.Y.; Chen, T.H.; Li, Y.H.; Chiu, K.P.; Nai, Y.S. Transcriptome-level assessment of the impact of deformed wing virus on honey bee larvae. Sci. Rep. 2021, 11, 15028. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.J.; Hansen, M.; Troemel, E. Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 2018, 14, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Taylor, H.E.; Dimopoulos, G. AgDscam, a hypervariable immunoglobulin domain-containing receptor of the Anopheles gambiae innate immune system. PLoS Biol. 2006, 4, e229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, K.; Koyama, H.; Ito, W.; Minakuchi, C.; Tanaka, T.; Miura, K. Involvement of NF-κB transcription factors in antimicrobial peptide gene induction in the red flour beetle, Tribolium castaneum. Dev. Comp. Immunol. 2012, 38, 342–351. [Google Scholar] [CrossRef]
- Moy, R.H.; Cherry, S. Antimicrobial autophagy: A conserved innate immune response in Drosophila. J. Innate Immun. 2013, 5, 444–455. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Protein Sequence Dataset | H. Sapiens | M. Musculus | C. Elegans | D. Melanogaster | B. Mori | B. Terrestris | N. Vitripennis | UniGene | Pfam |
|---|---|---|---|---|---|---|---|---|---|
| No. annotated predicted amino acid sequences (total 194,174) | 121,027 | 115,617 | 91,859 | 97,544 | 121,119 | 113,306 | 110,591 | 127,386 | 107,936 |
| % annotated sequences | 62.33% | 59.54% | 47.30% | 50.24% | 62.38% | 58.35% | 56.95% | 65.60% | 55.59% |
| Bioproject | Sample | Treatments | Reference | Sequence Read Archive Accession ID (Sample (Abbreviation) | Experimental Abbreviation) |
|---|---|---|---|---|---|
| PRJNA52851 | 4th instar honeybee larvae | Paenibacillus larvae orally inoculated and treated larvae were incubated 72 h. | [28] | SRR068395 (infected), SRR068396 (Control). | AFB |
| PRJEB6511 | Newly hatched larvae | Sacbrood virus (SBV)-deformed wing virus (DWV) mixture orally inoculated and treated larvae were incubated 9 d. Control was PBS. | [29] | ERR528750 (control_1_D1), ERR528751 (control_2_D2), ERR528753 (DWV_SBV_1_C5), ERR528754 (DWV_SBV_2_C6), | SBV_DWV |
| PRJNA445764 | Abdomens of worker adults | 1. Diet, Pollen (P), Bee-Pro (B), MagaBee (M) or control (carbohydrate diet) (C) fed 7 d. 2. Nosema apis and sucrose provided. 3. Each diet fed 7 d. | [30] | SRR6901841 (B2), SRR6901842 (B1), SRR6901848 (B3), SRR6901856 (BN1), SRR6901857 (BN2), SRR6901858 (BN3) SRR6901843 (P1), SRR6901844 (P2), SRR6901849 (P3), SRR6901859 (PN1), SRR6901860 (PN2), SRR6901862 (PN3) SRR6901845 (M1), SRR6901846 (M2), SRR6901847 (M3), SRR6901853 (MN1) SRR6901854 (MN2) SRR6901855 (MN3) SRR6901850 (C1) SRR6901851 (C2) SRR6901852 (C3) SRR6901861 (CN1) SRR6901863 (CN2) SRR6901864 (CN3) | B_BN (control vs. N. apis-inoculated samples reared with Bee-Pro) P_PN (control vs. N. apis-inoculated samples reared with pollen) M_MN (control vs. N. apis-inoculated samples reared with MagaBee) C_CN (control vs. N. apis-inoculated samples reared with carbohydrate diet) |
| PRJNA498919 | Whole bodies of adult bees | Samples reared with chestnut or rockrose (less nutritious), fed control or inoculated with Israeli acute paralysis virus (IAPV) containing 30% sucrose solution for 36 h | [31] | SRR8121077, SRR8121078 (CC_1), SRR8121079, SRR8121080 (CC_2), SRR8121081, SRR8121082 (CC_3), SRR8121083, SRR8121084 (CC_4), SRR8121085, SRR8121086 (CC_5), SRR8121087, SRR8121088 (CC_6), SRR8121101, SRR8121102 (CV_1), SRR8121103, SRR8121104 (CV_2), SRR8121105, SRR8121106 (CV_3), SRR8121107, SRR8121108 (CV_4), SRR8121109, SRR8121110 (CV_5), SRR8121111, SRR8121112 (CV_6) SRR8121089, SRR8121090 (RC_1), SRR8121091, SRR8121092 (RC_2), SRR8121093, SRR8121094 (RC_3), SRR8121095, SRR8121096 (RC_4), SRR8121097, SRR8121098 (RC_5), SRR8121099, SRR8121100 (RC_6), SRR8121113, SRR8121114 (RV_1), SRR8121115, SRR8121116 (RV_2), SRR8121117, SRR8121118 (RV_3), SRR8121119, SRR8121120 (RV_4), SRR8121121, SRR8121122 (RV_5), SRR8121123, SRR8121124 (RV_6) | CC_CV (control vs. IAPV-inoculated samples reared with chestnut) * RC_RV (control vs. IAPV-inoculated samples reared with rockrose) * |
| RJNA669279 | Larvae | DWV containing PBS (or OBS as control) were inoculated 12 h in larvae reared for 3 d from 1-d larvae. Treated larvae were incubated 4 d. | [32] | SRR12830831 (control_1), SRR14424854 (control_2), SRR14424852 (control_3), SRR12830832 (DWV_1), SRR12830855 (DWV_2), SRR12830853 (DWV_3) | DWV (control vs. DWV-infected) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoi, K.; Wakamiya, T.; Bono, H. Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data. Insects 2022, 13, 931. https://doi.org/10.3390/insects13100931
Yokoi K, Wakamiya T, Bono H. Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data. Insects. 2022; 13(10):931. https://doi.org/10.3390/insects13100931
Chicago/Turabian StyleYokoi, Kakeru, Takeshi Wakamiya, and Hidemasa Bono. 2022. "Meta-Analysis of the Public RNA-Seq Data of the Western Honeybee Apis mellifera to Construct Reference Transcriptome Data" Insects 13, no. 10: 931. https://doi.org/10.3390/insects13100931