
A Genetic Compensation Phenomenon and Global Gene Expression Changes in Sex-miR-2766-3p Knockout Strain of Spodoptera exigua Hübner (Lepidoptera: Noctuidae)

 ,
,  and
and
Abstract
:Simple Summary
Abstract
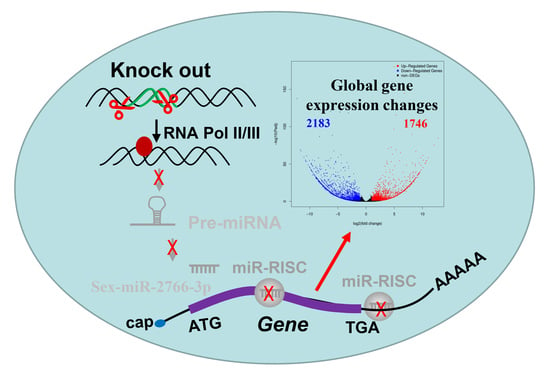
1. Introduction
2. Experimental Procedures
2.1. Insects and Cell Lines
2.2. Dual-Luciferase Reporter (DLR) Assay
2.3. sgRNA Production
2.4. Embryo Microinjection
2.5. CRISPR/Cas9 Knockout of Sex-miR-2766-3p
2.6. RNA Sequencing
2.7. RNA Extraction and Quantitative RT-PCR
3. Results
3.1. Sex-miR-2766-3p Regulates the Expression of SeCncC In Vitro
3.2. CRISPR/Cas9-Mediated Knockout of Sex-miRNA-2766-3p
3.3. Effect of Sex-miR-2766-3p Knockout on the Expression of the SeCncC Gene
3.4. Effect of Sex-miR-2766-3p Knockout on Global Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gen lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Yi, S.; Gao, Z.X.; Zhao, H.; Zeng, C.; Luo, W.; Chen, B.; Wang, W.-M. Identification and characterization of microRNAs involved in growth of blunt snout bream (Megalobrama amblycephala) by Solexa sequencing. BMC Genom. 2013, 14, 754. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Y.; Jagadeeswaran, G.; Ren, R.; Sunkar, R.; Jiang, H. Identification and developmental profiling of conserved and novel microRNAs in Manduca sexta. Insect Biochem. Mol. 2012, 42, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Zhang, Y.; Jiang, J.; Zhong, Y.; Yang, X.; Li, Z.; Huang, Y.; Tan, A. Identification of microRNAs in Helicoverpa armigera and Spodoptera litura based on deep sequencing and homology analysis. Int. J. Biol. Sci. 2013, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.J.; Zhu, F.; Liu, Y.J.; Li, Z.; Moural, T.W.; Liu, X.M.; Liu, X. MicroRNAs miR-14 and miR-2766 regulate tyrosine hydroxylase to control larval-pupal metamorphosis in Helicoverpa armigera. Pest Manag. Sci. 2022, 78, 3540–3550. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wang, A.; Duan, A.; Xue, C.; Wang, K.; Zhao, M.; Zhang, J. Four MicroRNAs, miR-13b-3p, miR-278-5p, miR-10483-5p, and miR-10485-5p, mediate insecticide tolerance in Spodoptera frugiperda. Front. Genet. 2022, 12, 820778. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Beumer, K.J.; Trautman, J.K.; Bozas, A.; Liu, J.L.; Rutter, J.; Gall, J.G.; Carroll, D. Efficient gene targeting in Drosophila by direct embryo injection with zincfinger nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 19821–19826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beumer, K.J.; Trautman, J.K.; Mukherjee, K.; Carroll, D. Donor DNA utilization during gene targeting with zinc-finger nucleases. G3-Genes. Genom. Genet. 2013, 3, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Shi, Y.; Wu, J.N.; Li, H.; Smagghe, G.; Liu, T.X. CRISPR/Cas9 in lepidopteran insects: Progress, application and prospects. J. Insect Physiol. 2021, 135, 104325. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.Y.; Wang, H.; Xu, Y.J.; Huang, J.L.; Wu, S.W.; Wu, Y.D.; Yang, Y. CRISPR/Cas9 mediated G4946E substitution in the ryanodine receptor of Spodoptera exigua confers high levels of resistance to diamide insecticides. Insect Biochem. Molec. Biol. 2017, 89, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.Y.; Xue, Y.X.; Wang, Z.Y.; Ren, X.; Aioub, A.A.; Wu, Y.D.; Yang, Y.H.; Hu, Z.N. Knockin of the G275E mutation of the nicotinic acetylcholine receptor (nAChR) α6 confers high levels of resistance to spinosyns in Spodoptera exigua. Insect Sci. 2022, 29, 478–486. [Google Scholar] [CrossRef]
- Zuo, Y.Y.; Xue, Y.J.; Lu, W.J.; Ma, H.H.; Chen, M.H.; Wu, Y.; Yang, Y.; Hu, Z. Functional validation of nicotinic acetylcholine receptor (nAChR) α6 as a target of spinosyns in Spodoptera exigua utilizing the CRISPR/Cas9 system. Pest Manag. Sci. 2020, 76, 2415–2422. [Google Scholar] [CrossRef]
- Zuo, Y.Y.; Shi, Y.; Zhang, F.; Guan, F.; Zhang, J.P.; Feyereisen, R.; Fabrick, J.A.; Yang, Y.; Wu, Y. Genome mapping coupled with CRISPR gene editing reveals a P450 gene confers avermectin resistance in the beet armyworm. PLoS Genet. 2021, 17, e1009680. [Google Scholar] [CrossRef]
- Berdegué, M.; Reitz, S.R.; Trumble, J.T. Host plant selection and development in Spodoptera exigua: Do mother and offspring know best? Entomol. Exp. Appl. 1998, 89, 57–64. [Google Scholar] [CrossRef]
- Che, W.; Shi, T.; Wu, Y.; Yang, Y. Insecticide resistance status of field populations of Spodoptera exigua (Lepidoptera: Noctuidae) from China. J. Econ. Entomol. 2013, 106, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Kalsi, M.; Palli, S.R. Cap n collar transcription factor regulates multiple genes coding for proteins involved in insecticide detoxification in the red flour beetle, Tribolium castaneum. Insect Biochem. Mol. Biol. 2017, 90, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yuxin, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2017, 7, gix120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.; Myers, E.W.; Lipman, D.J. Blast (basic local alignment search tool). Encycl. Genet. Genom. Proteom. Inform. 2008, 215, 221. [Google Scholar]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Park, C.Y.; Jeker, L.T.; Carver-Moore, K.; Oh, A.; Liu, H.J.; Cameron, R.; Richards, H.; Li, Z.; Adler, D.; Yoshinaga, Y.; et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep. 2012, 1, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Brolosy, M.A.; Stainier, D.Y. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meelad, M.D.; Kibibi, G.; Benjamin, E.P.; Hu, Y.; Styliani, M.; Albert, W.C.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem. Cell. 2011, 9, 166–175. [Google Scholar]
- Monique, N.O.; Katherine, H.S.; Zhang, Y.; Anne-Cécile, E.D.; Shuyun, R.J.; Scott, H.; Academia, E.C.; Shah, S.R.; Morton, J.F.; Holstein, C.A.; et al. The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l1. PLoS Genet. 2013, 9, e1003708. [Google Scholar]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, S.; Shi, Y.; Yang, Y.Y.; Wu, Y.D. Global gene expression changes induced by knockout of a protease gene cluster in Helicoverpa armigera with CRISPR/Cas9. J. Insect Physiol. 2020, 122, 104023. [Google Scholar] [CrossRef]
- Bouche, N.; Bouchez, D. Arabidopsis gene knockout: Phenotypes wanted. Curr. Opin. Plant Biol. 2001, 4, 111–117. [Google Scholar] [CrossRef]
- Rodriguez-Leal, D.; Xu, C.; Kwon, C.; Soyars, C.; Demesa-Arevalo, E.; Man, J.; Liu, L.; Lemmon, Z.H.; Jones, D.S.; Van Eck, J.; et al. Evolution of buffering in a genetic circuit controlling plant stem cell proliferation. Nat. Genet. 2019, 51, 786–792. [Google Scholar] [CrossRef]
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 2017, 21, 431–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambeyron, S.; Bickmore, W.A. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004, 18, 1119–1130. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Primer Sequence (5′–3′) | Purposes |
|---|---|---|
| 3UTR-MT-F | aacgagctcgctagcctcgagCCACCCAATGAACCGTACTTCCGACGATGATATGGACAGAAAAGCCAAGAGCTACGACCAGTGATAGGCGGTAACTACAAGCGCTCTACC | For making the mutated (MT) target sequence of CncC |
| 3UTR-MT-R | caggtcgactctagactcgagCAATTATTGCGTTGTCCAAGTCTTGTATGTGTGTATATGTATTAGGTAGAGCGCTTGTAGTTACCGCCTATCACTGGTCG | |
| 3UTR-WT-F | aacgagctcgctagcctcgagCCACCCAATGAACCGTACTTCCGACGATGATATGGACAGAAAAGCCAAGAGCTACGACCAGTGATTCCGCCTAACTAGTTCCCGAGATGG | For making the wild-type (WT) target sequence of CncC |
| 3UTR-WT-R | caggtcgactctagactcgagCAATTATTGCGTTGTCCAAGTCTTGTATGTGTGTATATGTATTACCATCTCGGGAACTAGTTAGGCGGAATCACTGGTCG | |
| sgRNAF1 | TAATACGACTCACTATAGCGGGTCGCGCGGAGCCCG | For making the DNA template of sgRNA1 |
| sgRNAR1 | TTCTAGCTCTAAAACCGGGCTCCGCGCGACCCG | |
| sgRNAF2 | TAATACGACTCACTATAGCTTCAGTCTTGTCGAATGG | For making the DNA template of sgRNA2 |
| sgRNAR2 | TTCTAGCTCTAAAACCCATTCGACAAGACTGAAG | |
| JC-miRNAF | CATGCTTACACAGTAGGAACGT | For detecting large fragment knockout |
| JC-miRNAR | GCGGAAGTTACTACACAAAGGG | |
| qmiR-2766F | AGTCTTGTCGAATGGTGGGT | For quantitative RT-PCR |
| qU6F | TTGGAACGATACAGAGAAGATTAGC | |
| qCncCF | ACAGAGCAATATTCCCAGTCCG | |
| qCncCR | AAGAACCACCATCTGACATGCT | |
| qGADPHF | AACATTTATCTCTACAACGCAATC | |
| qGADPHR | GTGACAACCACTCATCTATCTTC | |
| β-actinF | AGCGTGACATCAAGAGGACT | |
| β-actinR | CTCCATGTATGCCTGCTTCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, Y.; Wang, Z.; Ren, X.; Pei, Y.; Aioub, A.A.A.; Hu, Z. A Genetic Compensation Phenomenon and Global Gene Expression Changes in Sex-miR-2766-3p Knockout Strain of Spodoptera exigua Hübner (Lepidoptera: Noctuidae). Insects 2022, 13, 1075. https://doi.org/10.3390/insects13111075
Zuo Y, Wang Z, Ren X, Pei Y, Aioub AAA, Hu Z. A Genetic Compensation Phenomenon and Global Gene Expression Changes in Sex-miR-2766-3p Knockout Strain of Spodoptera exigua Hübner (Lepidoptera: Noctuidae). Insects. 2022; 13(11):1075. https://doi.org/10.3390/insects13111075
Chicago/Turabian StyleZuo, Yayun, Zeyu Wang, Xuan Ren, Yakun Pei, Ahmed A. A. Aioub, and Zhaonong Hu. 2022. "A Genetic Compensation Phenomenon and Global Gene Expression Changes in Sex-miR-2766-3p Knockout Strain of Spodoptera exigua Hübner (Lepidoptera: Noctuidae)" Insects 13, no. 11: 1075. https://doi.org/10.3390/insects13111075
APA StyleZuo, Y., Wang, Z., Ren, X., Pei, Y., Aioub, A. A. A., & Hu, Z. (2022). A Genetic Compensation Phenomenon and Global Gene Expression Changes in Sex-miR-2766-3p Knockout Strain of Spodoptera exigua Hübner (Lepidoptera: Noctuidae). Insects, 13(11), 1075. https://doi.org/10.3390/insects13111075