
Identifying the Genetic Distance Threshold for Entiminae (Coleoptera: Curculionidae) Species Delimitation via COI Barcodes



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
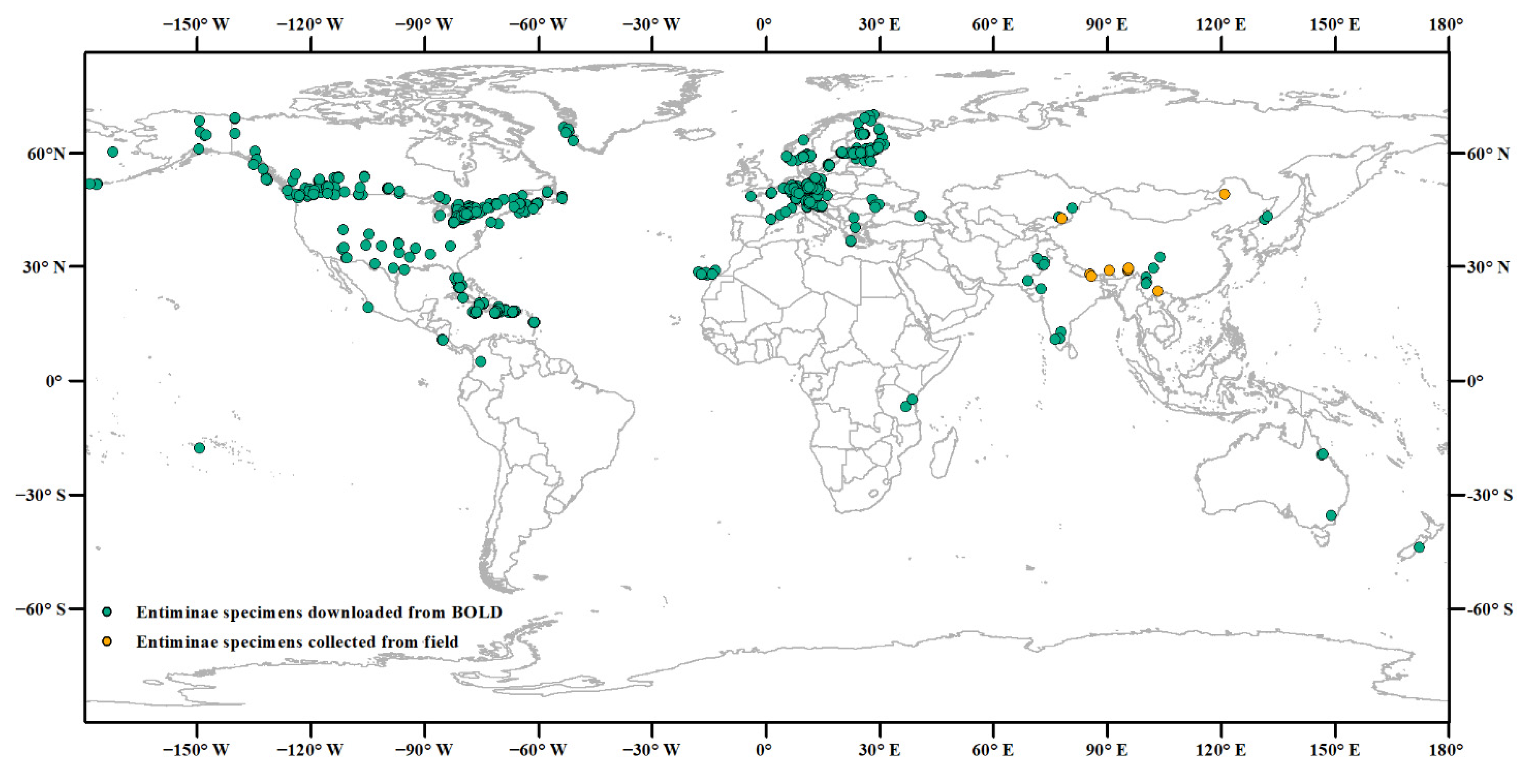
2.1. Publicly Available Data
2.2. Sample Collection and Identification
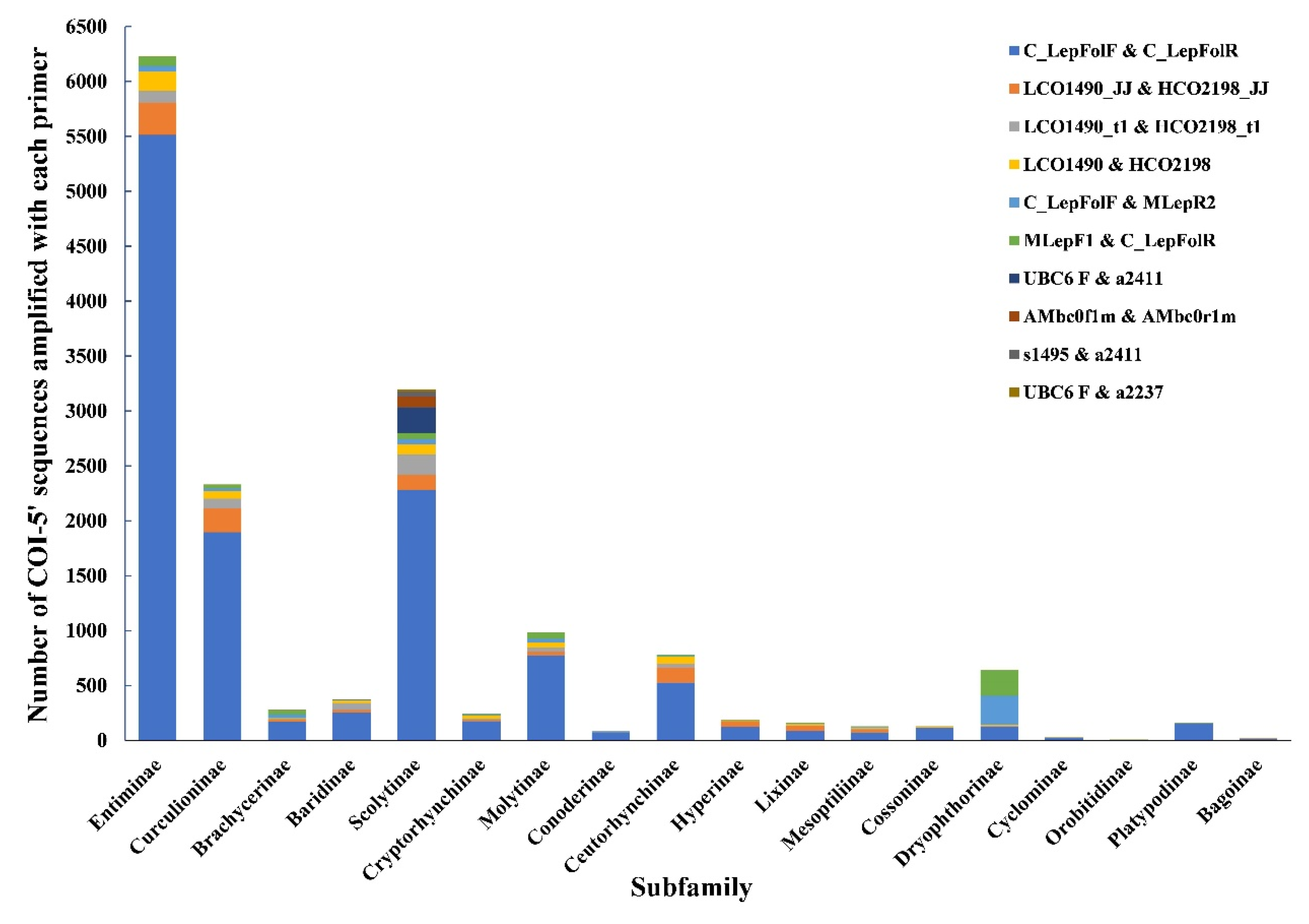
2.3. DNA Extraction, Amplification and DNA Sequencing
2.4. Sequence Analysis
3. Results
3.1. Downloading Data
3.2. Sequencing Data
3.3. Nucleotide Composition
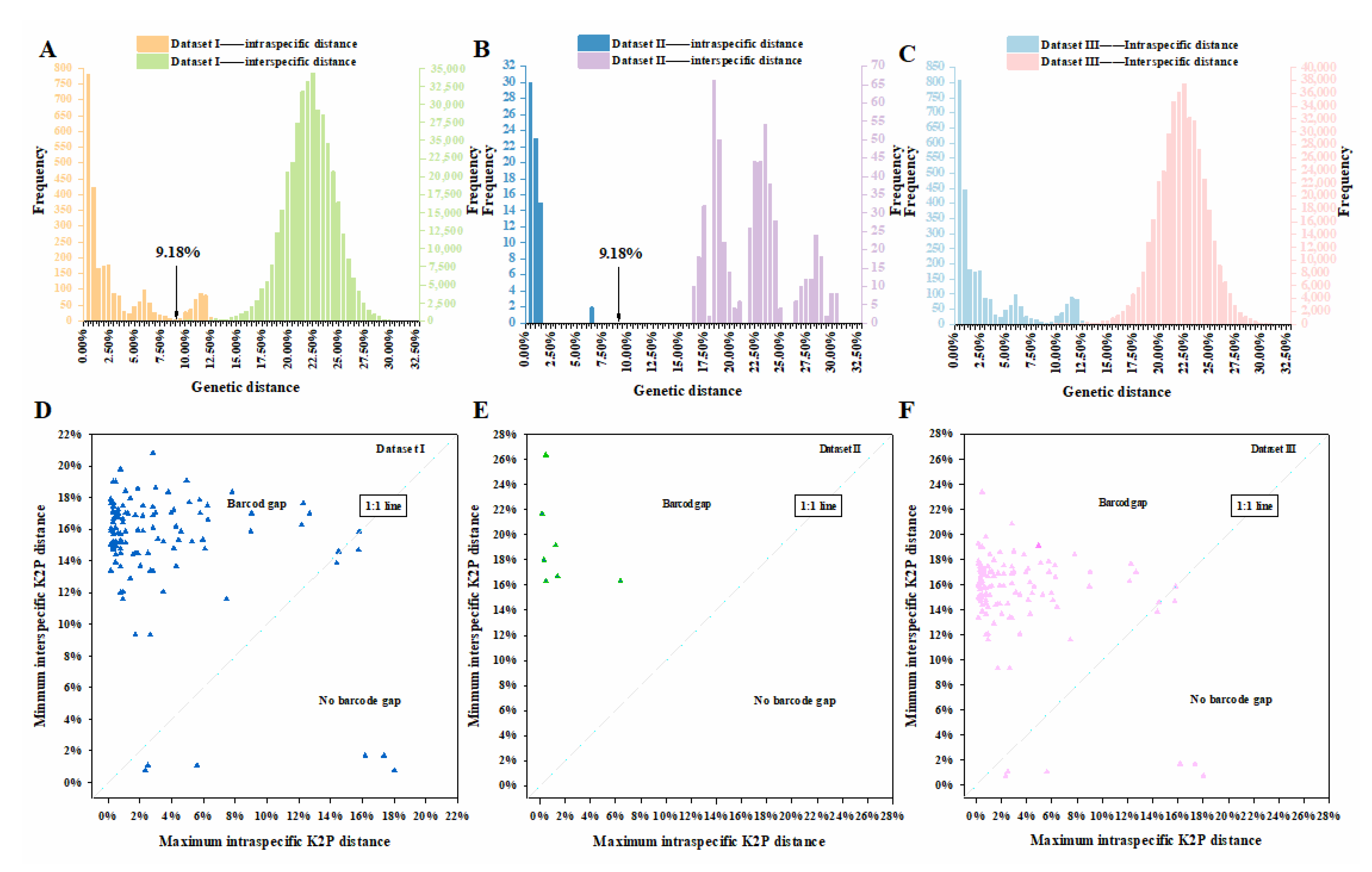
3.4. Genetic Distance
3.5. Barcode Gap Analysis
4. Discussion
4.1. The Choice of Primers
4.2. An Optimal Genetic Distance Threshold for Entiminae
4.3. High Intraspecific Divergence Versus Cryptic Species Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKenna, D.D.; Sequeira, A.S.; Marvaldi, A.E.; Farrell, B.D. Temporal lags and overlap in the diversification of weevils and flowering plants. Proc. Natl. Acad. Sci. USA 2009, 106, 7083–7088. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, P.; Bousquet, Y.; Davies, A.E.; Alonso-Zarazaga, M.A.; Lawrence, J.F.; Lyal, C.H.C.; Newton, A.F.; Reid, C.A.M.; Schmitt, M.; Ślipiński, S.A.; et al. Family-group names in Coleoptera (Insecta). Zookeys 2011, 881–972. [Google Scholar] [CrossRef] [Green Version]
- Oberprieler, R.G.; Marvaldi, A.E.; Anderson, R.S. Weevils, weevils, weevils everywhere. Zootaxa 2007, 1668, 491–520. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.; Addison, P.; Benoit, L.; Haran, J.M. Barcoding pest species in a biodiversity hot-spot: The South African polyphagous broad-nosed weevils (Coleoptera, Curculionidae, Entiminae). Biodivers. Data. J. 2021, 9, e66452. [Google Scholar] [CrossRef]
- Haran, J.M.; Hansen, S.; Benoit, L.; Addison, P. Description of five new species in the genus Phlyctinus Schoenherr (Coleoptera, Curculionidae): A first step in deciphering the P. callosus complex. Eur. J. Taxon. 2020, 669, 1–29. [Google Scholar] [CrossRef]
- Rodriguero, M.S.; Lanteri, A.A.; Confalonieri, V.A. Speciation in the asexual realm: Is the parthenogenetic weevil Naupactus cervinus a complex of species in statu nascendi? Mol. Phylogenet. Evol. 2013, 68, 644–656. [Google Scholar] [CrossRef]
- Germann, C.; Wyler, S.; Bernasconi, M.V. DNA barcoding of selected alpine beetles with focus on Curculionoidea (Coleoptera). Rev. Suisse Zool. 2020, 124, 15–38. [Google Scholar]
- Rosas, M.V.; Morrone, J.J.; del Río, M.G.; Lanteri, A.A. Phylogenetic analysis of the Pantomorus-Naupactus complex (Coleoptera: Curculionidae: Entiminae) from North and Central America. Zootaxa 2011, 2780, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Rannala, B.; Yang, Z.H. Species Delimitation; Scornavacca, C., Delsuc, F., Galtier, N., Eds.; No Commercial Publisher, Authors Open Access Book, 2020; Chapter No. 5.5; p. 5.5:3. [Google Scholar]
- Davis, J.I.; Nixon, K.C. Populations, genetic variation, and the delimitation of phylogenetic species. Syst. Biol. 1992, 41, 421–435. [Google Scholar] [CrossRef]
- Good, D.A.; Wake, D.B. Geographic variation and speciation in the torrent salamanders of the genus Rhyacotriton (Caudata: Rhyacotritonidae). Univ. Calif. Publ. Zool. 1992, 126, 1–91. [Google Scholar]
- Highton, R. Detecting Cryptic Species Using Allozyme Data. In The Biology of Plethodontid Salamanders; Bruce, R.C., Jaeger, R.G., Houck, L.D., Eds.; Springer: Boston, MA, USA, 2000; pp. 215–241. [Google Scholar]
- Porter, A.H. Testing nominal species boundaries using gene flow statistics: The taxonomy of two hybridizing admiral butterflies (Limenitis: Nymphalidae). Syst. Zool. 1990, 39, 131–147. [Google Scholar] [CrossRef]
- Grebennikov, V.V. Flightless Catapionus (Coleoptera: Curculionidae: Entiminae) in Southwest China survive the Holocene trapped on mountaintops: New species, unknown phylogeny and clogging taxonomy. Zootaxa 2016, 4205, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Cabras, A.A.; Cruz, R.D. DNA barcoding of selected Pachyrhynchus species (Coleoptera: Curculionidae) from Mt. Apo Natural Park, Philippines. Acta Biol. Univ. Daugavp. 2016, 16, 111–118. [Google Scholar]
- Schindel, D.E.; Miller, S.E. DNA barcoding a useful tool for taxonomists. Nature 2005, 435, 17. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, J.R.; Roe, A.D.; Sperling, F.A. Multi-locus species delimitation in closely related animals and fungi: One marker is not enough. Mol. Ecol. 2012, 21, 4422–4436. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.M.; Gonçalves, L.T. Borrowing the Pentatomomorpha tome from the DNA barcode library: Scanning the overall performance of cox1 as a tool. J. Zool. Syst. Evol. Res. 2021, 59, 992–1012. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2020, 21, 609–620. [Google Scholar] [CrossRef]
- Meyer, C.P.; Paulay, G. DNA Barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.G.; Lv, M.H.; Yi, W.B.; Zhu, W.B.; Bu, W.J. Species diversity can be overestimated by a fixed empirical threshold: Insights from DNA barcoding of the genus Cletus (Hemiptera: Coreidae) and the meta-analysis of COI data from previous phylogeographical studies. Mol. Ecol. Resour. 2017, 17, 314–323. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Tobias, J.A.; Seddon, N.; Spottiswoode, C.N.; Pilgrim, J.D.; Fishpool, L.D.; Collar, N.J. Quantitative criteria for species delimitation. Ibis 2010, 152, 724–746. [Google Scholar] [CrossRef]
- Meier, R.; Kwong, S.; Vaidya, G.; Ng, P.K.L. DNA Barcoding and Taxonomy in Diptera: A Tale of High Intraspecific Variability and Low Identification Success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgilio, M.; Jordaens, K.; Breman, F.C.; Backeljau, T.; De Meyer, M. Identifying insects with incomplete DNA barcode libraries, African fruit flies (Diptera: Tephritidae) as a test case. PLoS ONE 2012, 7, e31581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic. Acids. Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.C.; Chen, Y.Q. Economic Insect Fauna of China Fasc. 20 Coleoptera: Curculionidae (Ⅰ), 1st ed.; Science Press: Beijing, China, 1980; pp. 72–74. [Google Scholar]
- Ren, L. Systematics of the Leptomias Generic Group from China (Coleoptera: Curculionidae). Ph.D. Dissertation, Institute of Zoology, Chinese Academy of Science, Beijing, China, 2008. [Google Scholar]
- Hernández-Triana, L.M.; Prosser, S.W.; Rodríguez-Perez, M.A.; Chaverri, L.G.; Hebert, P.D.N.; Gregory, T.R. Recovery of DNA barcodes from blackfly museum specimens (Diptera: Simuliidae) using primer sets that target a variety of sequence lengths. Mol. Ecol. Resour. 2014, 14, 508–518. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; DeWaard, J.R.; Zakharov, E.V.; Prosser, S.W.; Sones, J.E.; McKeown, J.T.; Mantle, B.; La Salle, J. A DNA ‘Barcode Blitz’: Rapid digitization and sequencing of a natural history collection. PLoS ONE 2013, 8, e68535. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef] [Green Version]
- Burland, T.G. Dnastar’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Wiemers, M.; Fiedler, K. Does the DNA barcoding gap exist? —A case study in blue butterflies (Lepidoptera: Lycaenidae). Front. Zool. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.X.; Shi, L.C.; Han, J.P.; Li, G.; Lu, H.; Hou, J.Y.; Zhou, X.T.; Meng, F.Y.; Downie, S.R. Identification of species in the angiosperm family Apiaceae using DNA barcodes. Mol. Ecol. Resour. 2014, 14, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. A DNA-Based Registry for All Animal Species: The Barcode Index Number (BIN) System. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef] [Green Version]
- Gwiazdowski, R.A.; Foottit, R.G.; Maw, H.E.L.; Hebert, P.D. The Hemiptera (Insecta) of Canada: Constructing a Reference Library of DNA Barcodes. PLoS ONE 2015, 10, e0125635. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [Green Version]
- Albo, J.E.; Marelli, J.P.; Puig, A.S. Rapid Molecular Identification of Scolytinae (Coleoptera: Curculionidae). Int. J. Mol. Sci. 2019, 20, 5944. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, L.; Morinière, J.; Haszprunar, G.; Hebert, P.D.N.; Hausmann, A.; Köhler, F.; Balke, M. A comprehensive DNA barcode database for Central European beetles with a focus on Germany: Adding more than 3500 identified species to BOLD. Mol. Ecol. Resour. 2015, 15, 795–818. [Google Scholar] [CrossRef]
- Huang, W.; Xie, X.; Huo, L.; Liang, X.; Wang, X.; Chen, X. An integrative DNA barcoding framework of ladybird beetles (Coleoptera: Coccinellidae). Sci. Rep. 2020, 10, 10063. [Google Scholar] [CrossRef]
- D’Ercole, J.; Dincă, V.; Opler, P.A.; Kondla, N.; Schmidt, C.; Phillips, J.D.; Robbins, R.; Burns, J.M.; Miller, S.E.; Grishin, N.; et al. DNA barcode library for the butterflies of North America. PeerJ 2021, 9, e11157. [Google Scholar] [CrossRef]
- Ge, Y.; Xia, C.; Wang, J.; Zhang, X.; Ma, X.; Zhou, Q. The efficacy of DNA barcoding in the classification, genetic differentiation, and biodiversity assessment of benthic macroinvertebrates. Ecol. Evol. 2021, 11, 5669–5681. [Google Scholar] [CrossRef] [PubMed]
- Astrin, J.J.; Stüben, P.E.; Misof, B.; Wägele, J.W.; Gimnich, F.; Raupach, M.J.; Ahrens, D. Exploring diversity in cryptorhynchine weevils (Coleoptera) using distance-, character- and tree-based species delineation. Mol. Phylogenet. Evol. 2012, 63, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Zhang, G.; Ali, F. The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst. Biol. 2008, 57, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A.; Tao, R. Discordance of species trees with their most likely gene trees: The case of five taxa. Syst. Biol. 2008, 57, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, T.L.; Dawson, R.D.; Magalon, H.; Baudry, E. DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc. R. Soc. B 2007, 274, 1731–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Technique System for Molecular Identification of Quarantine Fruit Flies in China. Ph.D. Dissertation, China Agriculture University, Beijing, China, 2015. [Google Scholar]
- Park, D.S.; Foottit, R.; Maw, E.; Hebert, P.D.N. Barcoding bugs: DNA-based identification of the true bugs (Insecta: Hemiptera: Heteroptera). PLoS ONE 2011, 6, e18749. [Google Scholar] [CrossRef]
- Pentinsaari, M.; Hebert, P.D.N.; Mutanen, M. Barcoding beetles: A regional survey of 1872 species reveals high identification success and unusually deep interspecific divergences. PLoS ONE 2014, 9, e108651. [Google Scholar]
- Ascunce, M.S.; Nigg, H.N.; Clark, A. Molecular identification of the economically important invasive citrus root weevil Diaprepes abbreviatus (Coleoptera: Curculionidae). Fla. Entomol. 2009, 92, 167–172. [Google Scholar] [CrossRef]
- Przybycień, M.; Lachowska-Cierlik, D.; Wacławik, B.; Sprick, P.; Knutelski, S. The species status of the Otiorhynchus clavipes (Bonsdorff, 1785) species group (Coleoptera: Curculionidae): An integrative approach using molecular, morphological, ecological, and biogeographical data. Bonn Zoo. Bull. 2021, 70, 115–139. [Google Scholar]
- Tamar, K.; Carranza, S.; den Bosch, H.I.; Sindaco, R.; Moravec, J.; Meiri, S. Hidden relationships and genetic diversity: Molecular phylogeny and phylogeography of the Levantine lizards of the genus Phoenicolacerta (Squamata: Lacertidae). Mol. Phylogenet. Evol. 2015, 91, 86–97. [Google Scholar] [CrossRef]
- Brower, A.V.Z. Problems with DNA barcodes for species delimitation: ‘ten species’ of Astraptes fulgerator reassessed (Lepidoptera: Hesperiidae). Syst. Biodivers. 2006, 4, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.L.; Rosas-Puchuri, U.; Cañedo, R.M.; Alfaro-Shigueto, J.; Ayon, P.; Zelada-Mázmela, E.; Siccha-Ramirez, R.; Velez-Zuazo, X. DNA barcoding in the Southeast Pacific marine realm: Low coverage and geographic representation despite high diversity. PLoS ONE 2020, 15, e0244323. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, J.W.O. When one is not enough: Introgression of mitochondrial DNA in Drosophila. Mol. Biol. Evol. 2000, 17, 1126–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, J.C.; Alda, F.; Breitman, M.F.; Bermingham, E.; van den Berghe, E.P.; Johnson, J.B. Assessing Species Boundaries Using Multilocus Species Delimitation in a Morphologically Conserved Group of Neotropical Freshwater Fishes, the Poecilia sphenops Species Complex (Poeciliidae). PLoS ONE 2015, 10, e0121139. [Google Scholar] [CrossRef] [Green Version]
- Meiklejohn, K.A.; Damaso, N.; Robertson, J.M. Assessment of BOLD and GenBank-Their accuracy and reliability for the identification of biological materials. PLoS ONE 2019, 14, e0217084. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.M.; Janzen, D.H.; Hajibabaei, M.; Hallwachs, W.; Hebert, P.D. DNA barcodes of closely related (but morphologically and ecologically distinct) species of skipper butterflies (Hesperiidae) can differ by only one to three nucleotides. J. Lepid. Soc. 2007, 61, 138–153. [Google Scholar]
- Puillandre, N.; Modica, M.V.; Zhang, Y.; Sirovich, L.; Boisselier, M.C.; Cruaud, C.; Holford, M.; Samadi, S. Large-scale species delimitation method for hyperdiverse groups. Mol. Ecol. 2012, 21, 2671–2691. [Google Scholar] [CrossRef]
- Song, C.; Lin, X.L.; Wang, Q.; Wang, X.H. DNA barcodes successfully delimit morphospecies in a superdiverse insect genus. Zool. Scr. 2018, 47, 311–324. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5’ to 3’) | Reference | Notes |
|---|---|---|---|
| C_LepFolF (LepF1:LCO1490) | LCO1490 GGTCAACAAATCATAAAGATATTGG LepF1 ATTCAACCAATCATAAAGATATTGG | Hernández-Triana et al., 2014 | Cocktail Primer |
| C_LepFolR (LepR1:HCO2198) | HCO2198 TAAACTTCAGGGTGACCAAAAAATCA LepR1 TAAACTTCTGGATGTCCAAAAAATCA | Hernández-Triana et al., 2014 | Cocktail Primer |
| MLepF1 | GCTTTCCCACGAATAAATAATA | Hajibabaei et al., 2006 | |
| MLepR2 | GTTCAWCCWGTWCCWGCYCCATTTTC | Hajibabaei et al., 2006 |
| Primer Set | Length | PCR Success Rate |
|---|---|---|
| C_LepFolF + C_LepFolR | 658bp | 61.1% (33/54) |
| C_LepFolF + MLepR2 | 307bp | 71.4% (15/21) |
| MLepF1 + C_LepFolR | 407bp | 81.0% (17/21) |
| Nucleotide Position | Base Number (bp) | Conserved Site | Variable Sites | Parsim–Infor Sites | Singleton Sites | T (%) | C (%) | A (%) | G (%) | AT (%) | CG (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| The first position | 219 | 97 | 122 | 108 | 14 | 24.0 | 18.0 | 30.5 | 27.6 | 54.5 | 45.6 |
| The second position | 219 | 158 | 61 | 50 | 11 | 42.7 | 25.5 | 15.1 | 16.7 | 57.8 | 42.2 |
| The third position | 220 | 0 | 220 | 220 | 0 | 39.4 | 13.4 | 43.1 | 4.2 | 82.5 | 17.6 |
| All | 658 | 255 | 403 | 378 | 25 | 35.4 | 18.9 | 29.6 | 16.1 | 65.0 | 35.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; Ren, J.; Zhang, R. Identifying the Genetic Distance Threshold for Entiminae (Coleoptera: Curculionidae) Species Delimitation via COI Barcodes. Insects 2022, 13, 261. https://doi.org/10.3390/insects13030261
Ma Z, Ren J, Zhang R. Identifying the Genetic Distance Threshold for Entiminae (Coleoptera: Curculionidae) Species Delimitation via COI Barcodes. Insects. 2022; 13(3):261. https://doi.org/10.3390/insects13030261
Chicago/Turabian StyleMa, Zhuo, Jinliang Ren, and Runzhi Zhang. 2022. "Identifying the Genetic Distance Threshold for Entiminae (Coleoptera: Curculionidae) Species Delimitation via COI Barcodes" Insects 13, no. 3: 261. https://doi.org/10.3390/insects13030261
APA StyleMa, Z., Ren, J., & Zhang, R. (2022). Identifying the Genetic Distance Threshold for Entiminae (Coleoptera: Curculionidae) Species Delimitation via COI Barcodes. Insects, 13(3), 261. https://doi.org/10.3390/insects13030261