
Shallow Whole-Genome Sequencing of Aedes japonicus and Aedes koreicus from Italy and an Updated Picture of Their Evolution Based on Mitogenomics and Barcoding
 ,
,  ,
,  , , ,
, , ,  , , ,
, , , 
 and
and 
Abstract
:Simple Summary
Abstract
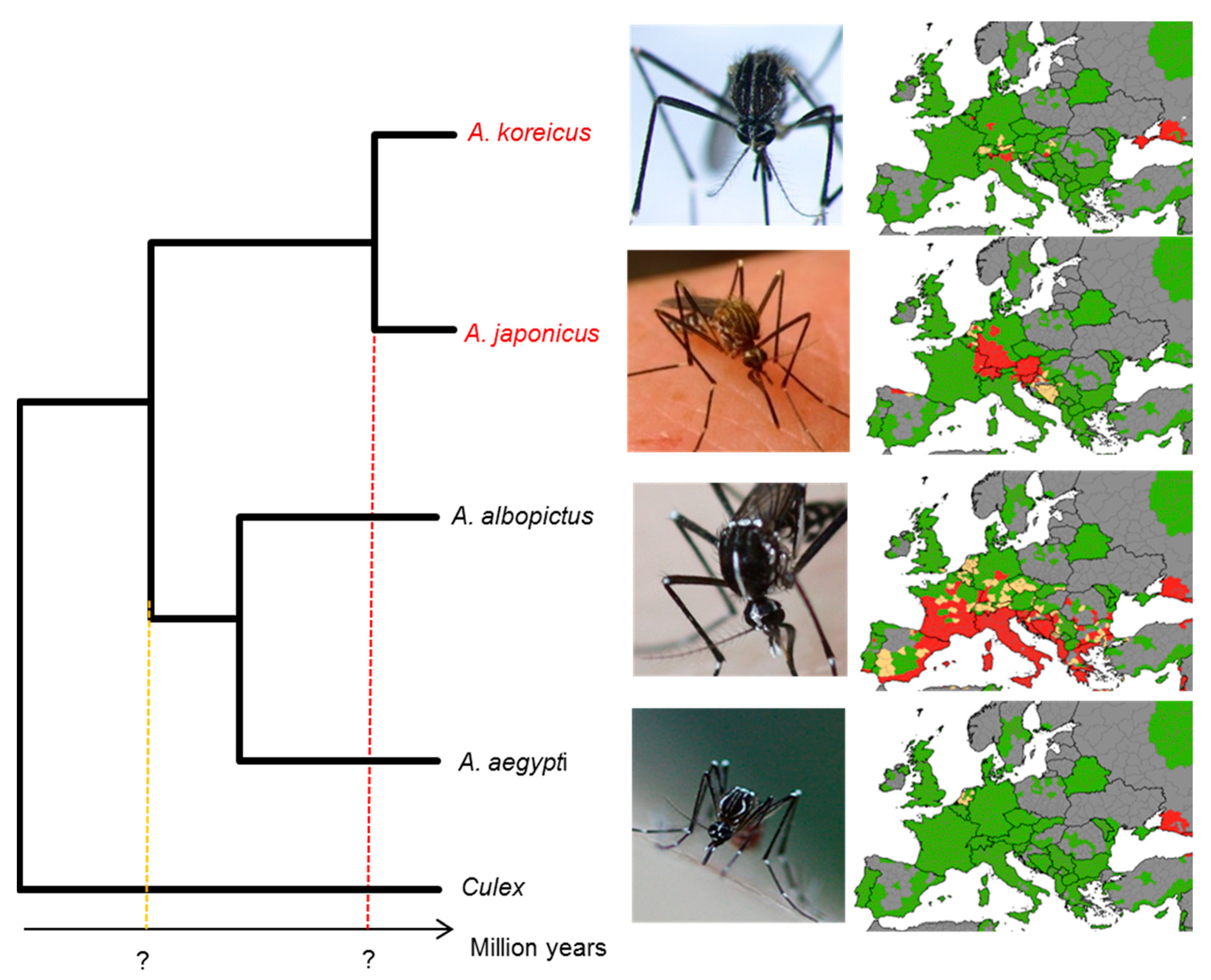
1. Introduction
2. Methods
2.1. Sampling, Sequencing, and Assembly of A. japonicus and A. koreicus Genomes and Mitogenomes
2.2. Microbial and Viral Profiling
2.3. Phylogenomic Datasets
2.4. Mitogenomic Datasets and Molecular Clock Analyses
2.5. Barcoding
3. Results and Discussion
3.1. Genome Skimming of A. japonicus and A. koreicus
3.2. Aedes Mitogenomes Displayed a Deeply Conserved Structure
3.3. Different Microbial Profiles, High Presence of Delftia, and Absence of Wolbachia
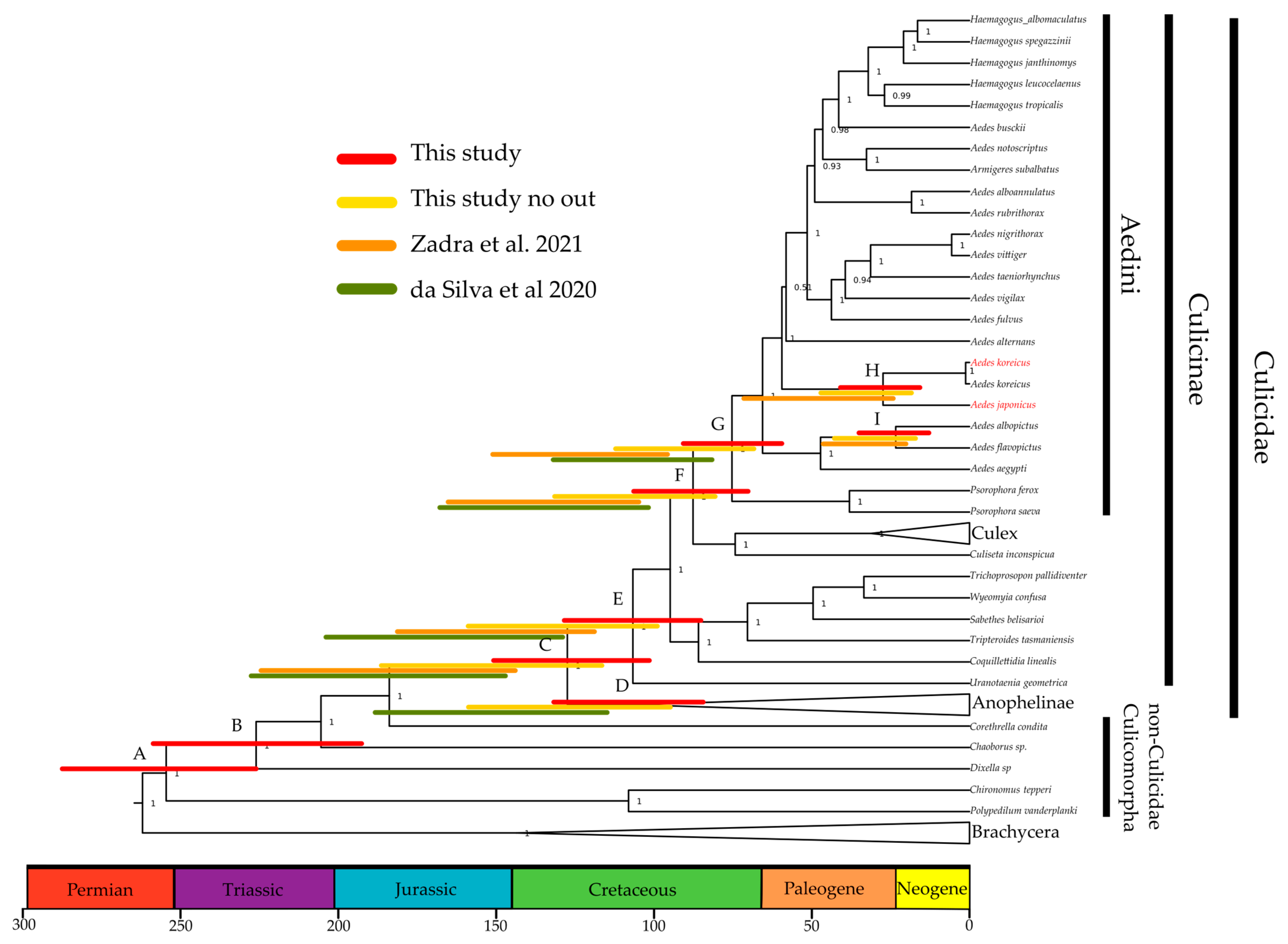
3.4. A More-Recent Timing of A. japonicus and A. koreicus Divergence Based on Mitogenomics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Node | Taxonomic Level | This Study | This Study Only Culicidae | Zadra et al. (2021) 1 | da Silva et al. (2020) 2 | Lorenz et al. (2021) 3 |
|---|---|---|---|---|---|---|
| A | Culicomorpha | 255 (226–287) | ||||
| B | Culicoidea | 226 (192–258) | 220 | |||
| C | Culicidae | 127 (101–150) | 151 (115–186) | 182 (143–223) | 182 (146–233) | 197 |
| D | Anophelinae | 106 (84–132) | 125 (94–159) | 145 (114–187) | 147 | |
| E | Culicinae | 106 (85–128) | 128 (98–158) | 150 (118–184) | 160 (128–205) | 153 |
| F | Culicini–Aedini split | 87 (70–106) | 105 (79–131) | 135 (104–164) | 130 (101–168) | 123 |
| G | Aedini | 75 (59–90) | 90 (67–113) | 111 (95–150) | 102 (81–132) | 74 |
| I | A. albopictus–A. flavipictus | 23 (12–34) | 29 (16–43) | 33 (20–46) | ||
| H | A. koreicus–A. japonicus | 27 (15–40) | 32 (18–48) | 46 (24–71) |
3.5. Outgroup-Rich Phylogenomics Provide a More-Recent Picture of Mosquito Radiations
3.6. A Large COI Barcode Analysis Supports Multiple Species within an Aedes japonicus + Aedes koreicus Species Complex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schaffner, F.; Medlock, J.M.; Van Bortel, W. Public health significance of invasive mosquitoes in Europe. Clin. Microbiol. Infect. 2013, 19, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Medlock, J.; Hansford, K.; Versteirt, V.; Cull, B.; Kampen, H.; Fontenille, D.; Hendrickx, G.; Zeller, H.; Van Bortel, W.; Schaffner, F. An entomological review of invasive mosquitoes in Europe. Bull. Entomol. Res. 2015, 105, 637–663. [Google Scholar] [CrossRef] [PubMed]
- Weger-Lucarelli, J.; Rückert, C.; Chotiwan, N.; Nguyen, C.; Luna, S.M.G.; Fauver, J.R.; Foy, B.D.; Perera, R.; Black, W.C.; Kading, R.C.; et al. Vector Competence of American Mosquitoes for Three Strains of Zika Virus. PLoS Negl. Trop. Dis. 2016, 10, e0005101. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.G.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.N.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.G.; Reiner, R.C., Jr.; Brady, O.J.; Messina, J.P.; Gilbert, M.; Pigott, D.M.; Yi, D.; Johnson, K.; Earl, L.; Marczak, L.B.; et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 2019, 4, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Weetman, D.; Kamgang, B.; Badolo, A.; Moyes, C.L.; Shearer, F.M.; Coulibaly, M.; Pinto, J.; Lambrechts, L.; McCall, P.J. Aedes Mosquitoes and Aedes-Borne Arboviruses in Africa: Current and Future Threats. Int. J. Environ. Res. Public Health 2018, 15, 220. [Google Scholar] [CrossRef]
- Rückert, C.; Ebel, G.D. How Do Virus–Mosquito Interactions Lead to Viral Emergence? Trends Parasitol. 2018, 34, 310–321. [Google Scholar] [CrossRef]
- Venter, M. Assessing the zoonotic potential of arboviruses of African origin. Curr. Opin. Virol. 2018, 28, 74–84. [Google Scholar] [CrossRef]
- Sacchetto, L.; Drumond, B.P.; Han, B.A.; Nogueira, M.L.; Vasilakis, N. Re-emergence of yellow fever in the neotropics—Quo vadis? Emerg. Top. Life Sci. 2020, 4, 411–422. [Google Scholar] [CrossRef]
- Epelboin, Y.; Talaga, S.; Epelboin, L.; Dusfour, I. Zika virus: An updated review of competent or naturally infected mosquitoes. PLoS Negl. Trop. Dis. 2017, 11, e0005933. [Google Scholar] [CrossRef]
- Sang, R.; Lutomiah, J.; Chepkorir, E.; Tchouassi, D.P. Evolving dynamics of Aedes-borne diseases in Africa: A cause for concern. Curr. Opin. Insect Sci. 2022, 53, 100958. [Google Scholar] [CrossRef]
- Medlock, J.M.; Hansford, K.M.; Schaffner, F.; Versteirt, V.; Hendrickx, G.; Zeller, H.; Van Bortel, W.; Swei, A.; Couper, L.I.; Coffey, L.L.; et al. A review of the invasive mosquitoes in Europe: Ecology, public health risks, and control options. Vector-Borne Zoonotic Dis. 2012, 12, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M.G.; Fonseca, D.M. Invasion biology of Aedes japonicus japonicus (Diptera: Culicidae). Annu. Rev. Entomol. 2014, 59, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.C.; Wilkerson, R.C.; Mogi, M.; Miyagi, I.; Toma, T.; Kim, H.-C.; Fonseca, D.M. Molecular phylogenetics of Aedes japonicus, a disease vector that recently invaded Western Europe, North America, and the Hawaiian Islands. J. Med. Entomol. 2010, 47, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Seidel, B.; Nowotny, N.; Bakonyi, T.; Allerberger, F.; Schaffner, F. Spread of Aedes japonicus japonicus (Theobald, 1901) in Austria, 2011-2015, and first records of the subspecies for Hungary, 2012, and the principality of Liechtenstein, 2015. Parasit Vectors 2016, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Seidel, B.; Montarsi, F.; Huemer, H.P.; Indra, A.; Capelli, G.; Allerberger, F.; Nowotny, N. First record of the Asian bush mosquito, Aedes japonicus japonicus, in Italy: Invasion from an established Austrian population. Parasites Vectors 2016, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.; Arnoldi, I.; Brilli, M.; Bandi, C.; Gabrieli, P.; Epis, S. Evidence for the spread of the alien species Aedes koreicus in the Lombardy region, Italy. Parasit Vectors 2021, 14, 534. [Google Scholar] [CrossRef] [PubMed]
- Arnoldi, I.; Negri, A.; Soresinetti, L.; Brambilla, M.; Carraretto, D.; Montarsi, F.; Roberto, P.; Mosca, A.; Rubolini, D.; Bandi, C.; et al. Assessing the distribution of invasive Asian mosquitoes in Northern Italy and modelling the potential spread of Aedes koreicus in Europe. Acta Trop. 2022, 232, 106536. [Google Scholar] [CrossRef]
- Capelli, G.; Drago, A.; Martini, S.; Montarsi, F.; Soppelsa, M.; Delai, N.; Ravagnan, S.; Mazzon, L.; Schaffner, F.; Mathis, A.; et al. First report in Italy of the exotic mosquito species Aedes (Finlaya) koreicus, a potential vector of arboviruses and filariae. Parasites Vectors 2011, 4, 188. [Google Scholar] [CrossRef]
- Huber, K.; Schuldt, K.; Rudolf, M.; Marklewitz, M.; Fonseca, D.M.; Kaufmann, C.; Tsuda, Y.; Junglen, S.; Krüger, A.; Becker, N.; et al. Distribution and genetic structure of Aedes japonicus japonicus populations (Diptera: Culicidae) in Germany. Parasitol. Res. 2014, 113, 3201–3210. [Google Scholar] [CrossRef]
- Medlock, J.M.; Hansford, K.M.; Vaux, A.G.C.; Cull, B.; Gillingham, E.; Leach, S. Assessment of the Public Health Threats Posed by Vector-Borne Disease in the United Kingdom (UK). Int. J. Environ. Res. Public Health 2018, 15, 2145. [Google Scholar] [CrossRef] [PubMed]
- Montarsi, F.; Drago, A.; Martini, S.; Calzolari, M.; De Filippo, F.; Bianchi, A.; Mazzucato, M.; Ciocchetta, S.; Arnoldi, D.; Baldacchino, F.; et al. Current distribution of the invasive mosquito species, Aedes koreicus [Hulecoeteomyia koreica] in northern Italy. Parasites Vectors 2015, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Bartlett-Healy, K.; Unlu, I.; Obenauer, P.; Hughes, T.; Healy, S.; Crepeau, T.; Farajollahi, A.; Kesavaraju, B.; Fonseca, D.; Schoeler, G.; et al. Larval mosquito habitat utilization and community dynamics of Aedes albopictus and Aedes japonicus (Diptera: Culicidae). J. Med. Entomol. 2012, 49, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, K.; Kiss, V.; Zana, B.; Schmieder, V.; Kepner, A.; Jakab, F.; Kemenesi, G. Emergence of Aedes koreicus (Diptera: Culicidae) in an urban area, Hungary, 2016. Parasitol. Res. 2016, 115, 4687–4689. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, K.; Zeghbib, S.; Arnoldi, D.; Marini, G.; Manica, M.; Michelutti, A.; Montarsi, F.; Deblauwe, I.; Van Bortel, W.; Smitz, N.; et al. Aedes koreicus, a vector on the rise: Pan-European genetic patterns, mitochondrial and draft genome sequencing. PLoS ONE 2022, 17, e0269880. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Jung, J. Comparative population genetics of the invasive mosquito Aedes albopictus and the native mosquito Aedes flavopictus in the Korean peninsula. Parasit Vectors 2021, 14, 377. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, D.M.; Widdel, A.K.; Hutchinson, M.; Spichiger, S.-E.; Kramer, L.D. Fine-scale spatial and temporal population genetics of Aedes japonicus, a new US mosquito, reveal multiple introductions. Mol. Ecol. 2010, 19, 1559–1572. [Google Scholar] [CrossRef]
- Smitz, N.; De Wolf, K.; Deblauwe, I.; Kampen, H.; Schaffner, F.; De Witte, J.; Schneider, A.; Verlé, I.; Vanslembrouck, A.; Dekoninck, W.; et al. Population genetic structure of the Asian bush mosquito, Aedes japonicus (Diptera, Culicidae), in Belgium suggests multiple introductions. Parasites Vectors 2021, 14, 179. [Google Scholar] [CrossRef]
- Baharmand, I.; Coatsworth, H.; Peach, D.A.H.; Belton, P.; Lowenberger, C. Molecular relationships of introduced Aedes japonicus (Diptera: Culicidae) populations in British Columbia, Canada using mitochondrial DNA. J. Vector Ecol. 2020, 45, 285–296. [Google Scholar] [CrossRef]
- Widdel, A.K.; McCuiston, L.J.; Crans, W.J.; Kramer, L.D.; Fonseca, D.M. Finding needles in the haystack: Single copy microsatellite loci for Aedes japonicus (Diptera: Culicidae). Am. J. Trop. Med. Hyg. 2005, 73, 744–748. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16222019 (accessed on 22 November 2023). [CrossRef]
- Seok, S.; Jacobsen, C.M.; Romero-Weaver, A.L.; Wang, X.; Nguyen, V.T.; Collier, T.C.; Riles, M.T.; Akbari, O.S.; Lee, Y. Complete mitogenome sequence of Aedes (Hulecoeteomyia) japonicus japonicus from Hawai’i Island. Mitochondrial DNA Part B 2023, 8, 64–68. [Google Scholar] [CrossRef]
- Soghigian, J.; Andreadis, T.G.; Livdahl, T.P. From ground pools to treeholes: Convergent evolution of habitat and phenotype in Aedes mosquitoes. BMC Evol. Biol. 2017, 17, 262. [Google Scholar] [CrossRef] [PubMed]
- Zadra, N.; Rizzoli, A.; Rota-Stabelli, O. Chronological Incongruences between Mitochondrial and Nuclear Phylogenies of Aedes Mosquitoes. Life 2021, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, A.; Ometto, L.; Rota-Stabelli, O. The complete mitogenome of the european mantis, mantis religiosa, from italy: Implications for the origin of north american mantis population. Bull. Insectology 2021, 74, 253–257. Available online: https://cris.unibo.it/handle/11585/849194 (accessed on 22 November 2023).
- Valerio, F.; Zadra, N.; Rota-Stabelli, O.; Ometto, L. The Impact of Fast Radiation on the Phylogeny of Bactrocera Fruit Flies as Revealed by Multiple Evolutionary Models and Mutation Rate-Calibrated Clock. Insects 2022, 13, 603. [Google Scholar] [CrossRef] [PubMed]
- Marini, G.; Arnoldi, D.; Baldacchino, F.; Capelli, G.; Guzzetta, G.; Merler, S.; Montarsi, F.; Rizzoli, A.; Rosà, R. First report of the influence of temperature on the bionomics and population dynamics of Aedes koreicus, a new invasive alien species in Europe. Parasites Vectors 2019, 12, 524. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Manghi, P.; Blanco-Míguez, A.; Manara, S.; NabiNejad, A.; Cumbo, F.; Beghini, F.; Armanini, F.; Golzato, D.; Huang, K.D.; Thomas, A.M.; et al. MetaPhlAn 4 profiling of unknown species-level genome bins improves the characterization of diet-associated microbiome changes in mice. Cell Rep. 2023, 42, 112464. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; on behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.F.; Machado, L.C.; de Paula, M.B.; Vieira, C.J.d.S.P.; Bronzoni, R.V.d.M.; Santos, M.A.V.d.M.; Wallau, G.L. Culicidae evolutionary history focusing on the Culicinae subfamily based on mitochondrial phylogenomics. Sci. Rep. 2020, 10, 18823. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Borkent, A.; Grimaldi, D.A. The Earliest Fossil Mosquito (Diptera: Culicidae), in Mid-Cretaceous Burmese Amber. Ann. Entomol. Soc. Am. 2004, 97, 882–888. [Google Scholar] [CrossRef]
- Benton, M.J.; Donoghue, P.C.J. Paleontological evidence to date the tree of life. Mol. Biol. Evol. 2007, 24, 26–53. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Poinar, G.; Zavortink, T.J.; Brown, A. Priscoculex burmanicus n. gen. et sp. (Diptera: Culicidae: Anophelinae) from mid-Cretaceous Myanmar amber. Hist. Biol. 2020, 32, 1157–1162. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Feuda, R.; Goulty, M.; Zadra, N.; Gasparetti, T.; Rosato, E.; Pisani, D.; Rizzoli, A.; Segata, N.; Ometto, L.; Stabelli, O.R. Phylogenomics of Opsin Genes in Diptera Reveals Lineage-Specific Events and Contrasting Evolutionary Dynamics in Anopheles and Drosophila. Genome Biol. Evol. 2021, 13, evab170. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.; Ometto, L.; Crava, C.M.; Revadi, S.; Kaur, R.; Horner, D.S.; Pisani, D.; Dekker, T.; Anfora, G.; Rota-Stabelli, O. The Evolution of Olfactory Gene Families in Drosophilaand the Genomic Basis of chemical-Ecological Adaptation in Drosophila suzukii. Genome Biol. Evol. 2016, 8, 2297–2311. [Google Scholar] [CrossRef]
- Richter, S.; Schwarz, F.; Hering, L.; Böggemann, M.; Bleidorn, C. The Utility of Genome Skimming for Phylogenomic Analyses as Demonstrated for Glycerid Relationships (Annelida, Glyceridae). Genome Biol. Evol. 2015, 7, 3443–3462. [Google Scholar] [CrossRef]
- Alfano, N.; Tagliapietra, V.; Rosso, F.; Manica, M.; Arnoldi, D.; Pindo, M.; Rizzoli, A. Changes in Microbiota Across Developmental Stages of Aedes koreicus, an Invasive Mosquito Vector in Europe: Indications for Microbiota-Based Control Strategies. Front. Microbiol. 2019, 10, 2832. [Google Scholar] [CrossRef]
- MacLeod, H.J.; Dimopoulos, G.; Short, S.M. Larval Diet Abundance Influences Size and Composition of the Midgut Microbiota of Aedes aegypti Mosquitoes. Front. Microbiol. 2021, 12, 645362. [Google Scholar] [CrossRef]
- Coon, K.L.; Brown, M.R.; Strand, M.R. Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae). Parasit. Vectors 2016, 9, 375. [Google Scholar] [CrossRef]
- Huang, W.; Rodrigues, J.; Bilgo, E.; Tormo, J.R.; Challenger, J.D.; De Cozar-Gallardo, C.; Pérez-Victoria, I.; Reyes, F.; Castañeda-Casado, P.; Gnambani, E.J.; et al. Delftia tsuruhatensis TC1 symbiont suppresses malaria transmission by anopheline mosquitoes. Science 2023, 381, 533–540. [Google Scholar] [CrossRef]
- Coon, K.L.; Vogel, K.J.; Brown, M.R.; Strand, M.R. Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 2014, 23, 2727–2739. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, T.; Wu, Y.; Zhong, D.; Zhou, G.; Su, X.; Xu, J.; Sotero, C.F.; Sadruddin, A.A.; Wu, K.; et al. Bacterial microbiota assemblage in Aedes albopictus mosquitoes and its impacts on larval development. Mol. Ecol. 2018, 27, 2972–2985. [Google Scholar] [CrossRef] [PubMed]
- Möhlmann, T.W.R.; Vogels, C.B.F.; Göertz, G.P.; Pijlman, G.P.; Ter Braak, C.J.F.; Beest, D.E.T.; Hendriks, M.; Nijhuis, E.H.; Warris, S.; Drolet, B.S.; et al. Impact of Gut Bacteria on the Infection and Transmission of Pathogenic Arboviruses by Biting Midges and Mosquitoes. Microb. Ecol. 2020, 80, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Yabe, A.; Jeong, E.; Kim, K.; Uemura, K. Oligocene–Neogene fossil history of Asian endemic conifer genera in Japan and Korea. J. Syst. Evol. 2019, 57, 114–128. [Google Scholar] [CrossRef]
- Kim, H.-J.; Moon, S.; Kim, C.; Kim, K.-H.; Seo, W.; Cho, K.-H.; Moon, H.-J.; Lee, G.H. Neotectonics at the SE continental margin of the Korean peninsula: Implications for the back-arc region behind the SW japan arc. Pure Appl. Geophys. 2022, 179, 3945–3966. [Google Scholar] [CrossRef]
- O’brien, C.L.; Huber, M.; Thomas, E.; Pagani, M.; Super, J.R.; Elder, L.E.; Hull, P.M. The enigma of Oligocene climate and global surface temperature evolution. Proc. Natl. Acad. Sci. USA 2020, 117, 25302–25309. [Google Scholar] [CrossRef] [PubMed]
- Marini, G.; Manica, M.; Arnoldi, D.; Inama, E.; Rosà, R.; Rizzoli, A. Influence of Temperature on the Life-Cycle Dynamics of Aedes albopictus Population Established at Temperate Latitudes: A Laboratory Experiment. Insects 2020, 11, 808. [Google Scholar] [CrossRef]
- Reuss, F.; Wieser, A.; Niamir, A.; Bálint, M.; Kuch, U.; Pfenninger, M.; Müller, R. Thermal experiments with the Asian bush mosquito (Aedes japonicus japonicus) (Diptera: Culicidae) and implications for its distribution in Germany. Parasites Vectors 2018, 11, 81. [Google Scholar] [CrossRef]
- Lorenz, C.; Alves, J.M.P.; Foster, P.G.; Suesdek, L.; Sallum, M.A.M. Phylogeny and temporal diversification of mosquitoes (Diptera: Culicidae) with an emphasis on the Neotropical fauna. Syst. Entomol. 2021, 46, 798–811. [Google Scholar] [CrossRef]
- Chen, X.-G.; Jiang, X.; Gu, J.; Xu, M.; Wu, Y.; Deng, Y.; Zhang, C.; Bonizzoni, M.; Dermauw, W.; Vontas, J.; et al. Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E5907–E5915. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.E.R.; Schrago, C.G. The influence of taxon sampling on Bayesian divergence time inference under scenarios of rate heterogeneity among lineages. J. Theor. Biol. 2015, 364, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Spasojevic, T.; Broad, G.R.; Sääksjärvi, I.E.; Schwarz, M.; Ito, M.; Korenko, S.; Klopfstein, S. Mind the Outgroup and Bare Branches in Total-Evidence Dating: A Case Study of Pimpliform Darwin Wasps (Hymenoptera, Ichneumonidae). Syst. Biol. 2021, 70, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Bromham, L.; Duchêne, S.; Hua, X.; Ritchie, A.M.; Duchêne, D.A.; Ho, S.Y.W. Bayesian molecular dating: Opening up the black box. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1165–1191. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, I.; Lee, K.W. Morphology and biology of Aedes japonicus and Aedes koreicus observed in laboratory experiments. Jpn. J. Sanit. Zool. 1975, 25, 300. [Google Scholar]
- Versteirt, V.; Nagy, Z.T.; Roelants, P.; Denis, L.; Breman, F.C.; Damiens, D.; Dekoninck, W.; Backeljau, T.; Coosemans, M.; Van Bortel, W. Identification of Belgian mosquito species (Diptera: Culicidae) by DNA barcoding. Mol. Ecol. Resour. 2015, 15, 449–457. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zadra, N.; Tatti, A.; Silverj, A.; Piccinno, R.; Devilliers, J.; Lewis, C.; Arnoldi, D.; Montarsi, F.; Escuer, P.; Fusco, G.; et al. Shallow Whole-Genome Sequencing of Aedes japonicus and Aedes koreicus from Italy and an Updated Picture of Their Evolution Based on Mitogenomics and Barcoding. Insects 2023, 14, 904. https://doi.org/10.3390/insects14120904
Zadra N, Tatti A, Silverj A, Piccinno R, Devilliers J, Lewis C, Arnoldi D, Montarsi F, Escuer P, Fusco G, et al. Shallow Whole-Genome Sequencing of Aedes japonicus and Aedes koreicus from Italy and an Updated Picture of Their Evolution Based on Mitogenomics and Barcoding. Insects. 2023; 14(12):904. https://doi.org/10.3390/insects14120904
Chicago/Turabian StyleZadra, Nicola, Alessia Tatti, Andrea Silverj, Riccardo Piccinno, Julien Devilliers, Clifton Lewis, Daniele Arnoldi, Fabrizio Montarsi, Paula Escuer, Giuseppe Fusco, and et al. 2023. "Shallow Whole-Genome Sequencing of Aedes japonicus and Aedes koreicus from Italy and an Updated Picture of Their Evolution Based on Mitogenomics and Barcoding" Insects 14, no. 12: 904. https://doi.org/10.3390/insects14120904
APA StyleZadra, N., Tatti, A., Silverj, A., Piccinno, R., Devilliers, J., Lewis, C., Arnoldi, D., Montarsi, F., Escuer, P., Fusco, G., De Sanctis, V., Feuda, R., Sánchez-Gracia, A., Rizzoli, A., & Rota-Stabelli, O. (2023). Shallow Whole-Genome Sequencing of Aedes japonicus and Aedes koreicus from Italy and an Updated Picture of Their Evolution Based on Mitogenomics and Barcoding. Insects, 14(12), 904. https://doi.org/10.3390/insects14120904