
Nine Mitochondrial Genomes of Phasmatodea with Two Novel Mitochondrial Gene Rearrangements and Phylogeny

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling Collection
2.2. DNA Extraction and Sequencing
2.3. Mitogenome Assembly, Annotation and Sequence Analyses
2.4. Phylogenetic Analyses
3. Results
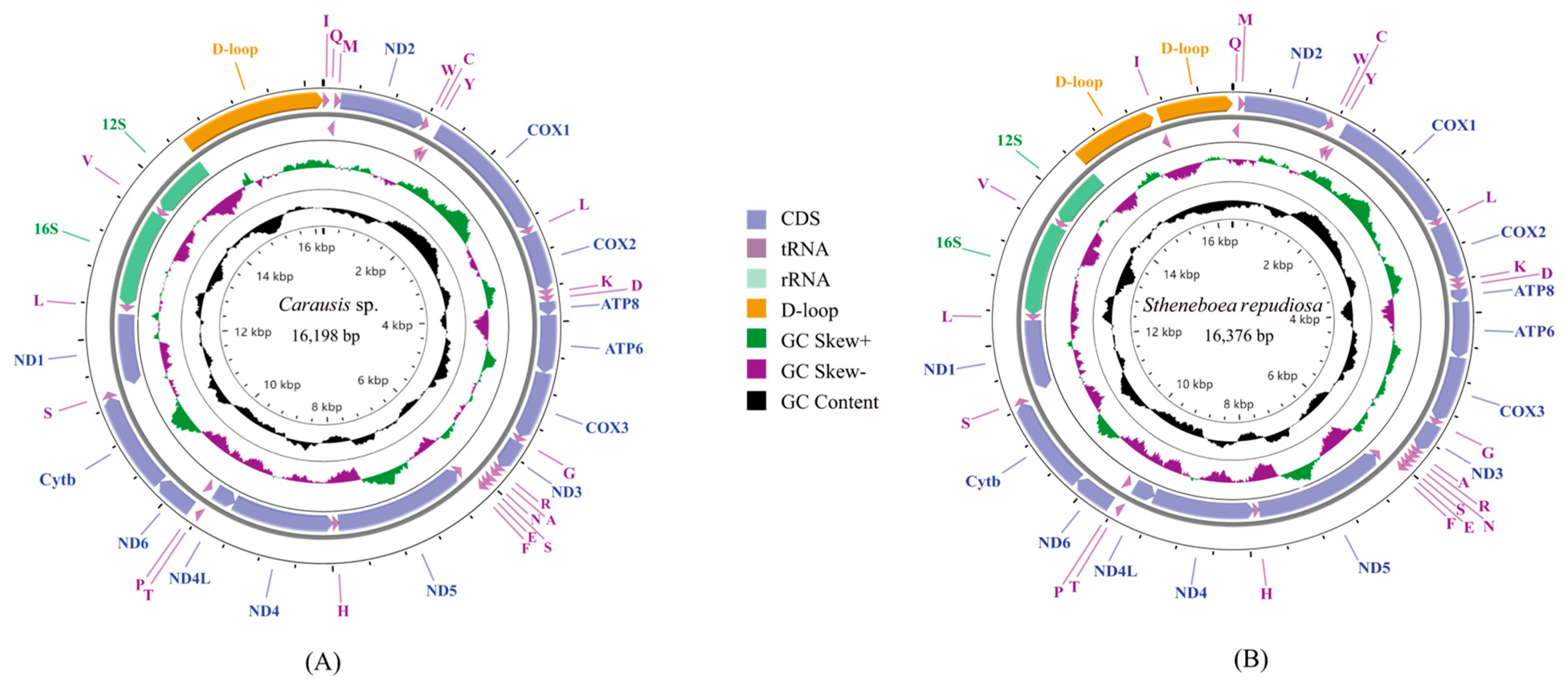
3.1. Basic Features of Mitogenomes and Gene Rearrangement
3.2. Protein-Coding Genes and Codon Usages
3.3. Transfer RNA and Ribosomal RNA Genes
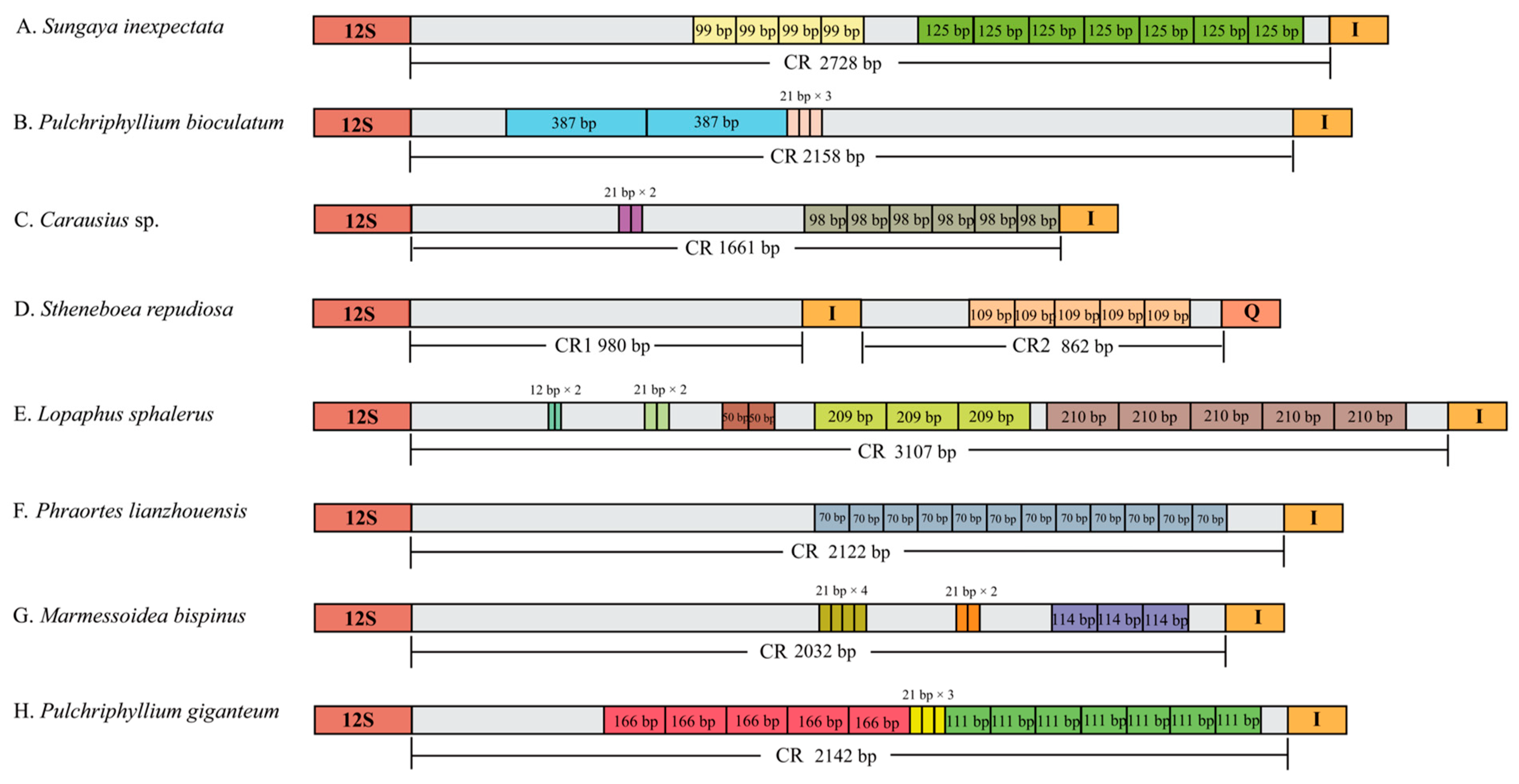
3.4. Control Region
3.5. Phylogenetic Analyses
4. Discussion
4.1. Non-Coding Regions and Control Regions of Mitogenomes
4.2. Gene Rearrangement
4.3. Phylogenetic Relationship within Phasmatodea
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bradler, S.; Buckley, T.R. Biodiversity of Phasmatodea. In Insect Biodiversity; Adler, P.H., Foottit, R.G., Eds.; John Wiley and Sons Ltd.: Hoboken, NJ, USA, 2018; pp. 281–313. [Google Scholar]
- Phasmida Species File Online. Version 5.0/5.0. Available online: http://Phasmida.SpeciesFile.org (accessed on 21 March 2023).
- Buckley, T.R.; Attanayake, D.; Bradler, S. Extreme convergence in stick insect evolution: Phylogenetic placement of the Lord Howe Island tree lobster. Proc. R. Soc. B 2009, 276, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Cumming, R.T.; Bank, S.; Tirant, S.L.; Bradler, S. Notes on the leaf insects of the genus Phyllium of Sumatra and Java, Indonesia, including the description of two new species with purple coxae (Phasmatodea, Phylliidae). ZooKeys 2020, 913, 89. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.A.; Bradler, S.; Whiting, M.F. Evolution of oviposition techniques in stick and leaf insects (Phasmatodea). Front. Ecol. Evol. 2018, 6, 216. [Google Scholar] [CrossRef]
- Bradler, S.; Cliquennois, N.; Buckley, T.R. Single origin of the Mascarene stick insects: Ancient radiation on sunken islands? BMC Evol. Biol. 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Brown, W.M.; George Jr, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Howe, K.; Chow, W.; Collins, J.; Pelan, S.; Pointon, D.L.; Sims, Y.; Torrance, J.; Tracey, A.; Wood, J. Significantly improving the quality of genome assemblies through curation. Gigascience 2021, 10, giaa153. [Google Scholar] [CrossRef]
- Hunt, M.; Silva, N.D.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef]
- Rokas, A.; Holland, P.W. Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol. 2000, 15, 454–459. [Google Scholar] [CrossRef]
- Yan, D.; Tang, Y.; Xue, X.; Wang, M.; Liu, F.; Fan, J. The complete mitochondrial genome sequence of the western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae) contains triplicate putative control regions. Gene 2012, 506, 117–124. [Google Scholar] [CrossRef]
- Tyagi, K.; Chakraborty, R.; Cameron, S.L.; Sweet, A.D.; Chandra, K.; Kumar, V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci. Rep. 2020, 10, 695. [Google Scholar] [CrossRef]
- Liu, Q.; He, J.; Song, F.; Tian, L.; Cai, W.; Li, H. Positive correlation of the gene rearrangements and evolutionary rates in the mitochondrial genomes of thrips (Insecta: Thysanoptera). Insects 2022, 13, 585. [Google Scholar] [CrossRef]
- Lin, Y.J.; Cai, L.N.; Zhao, Y.Y.; Cheng, H.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Novel Mitochondrial Gene rearrangement and intergenic regions exist in the mitochondrial genomes from four newly established ramilies of praying mantises (Insecta: Mantodea). Insects 2022, 13, 564. [Google Scholar] [CrossRef]
- Xu, X.D.; Guan, J.Y.; Zhang, Z.Y.; Cao, Y.R.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Novel tRNA gene rearrangements in the mitochondrial genomes of praying mantises (Mantodea: Mantidae): Translocation, duplication and pseudogenization. Int. J. Biol. Macromol. 2021, 185, 403–411. [Google Scholar] [CrossRef]
- Ye, F.; Lan, X.E.; Zhu, W.B.; You, P. Mitochondrial genomes of praying mantises (Dictyoptera, Mantodea): Rearrangement, duplication, and reassignment of tRNA genes. Sci. Rep. 2016, 6, 25634. [Google Scholar] [CrossRef]
- Zhang, L.P.; Cai, Y.Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. Gene characteristics of the complete mitochondrial genomes of Paratoxodera polyacantha and Toxodera hauseri (Mantodea: Toxoderidae). PeerJ 2018, 6, e4595. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, B.Y.; Zhu, J.C.; van Achterberg, C.; Tang, P.; Chen, X.X. The first two mitochondrial genomes of wood wasps (Hymenoptera: Symphyta): Novel gene rearrangements and higher-level phylogeny of the basal hymenopterans. Int. J. Biol. Macromol. 2019, 123, 1189–1196. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.; Wu, Y.; Song, F.; Cai, W.; Li, H. Novel tRNA gene rearrangements in the mitochondrial genome of Camarochiloides weiweii (Hemiptera: Pachynomidae). Int. J. Biol. Macromol. 2020, 165, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Shao, R.; Shi, A.; Bai, X.; Zheng, X.; Heiss, E.; Cai, W. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 2016, 6, 25725. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Song, H.; Cameron, S.L.; Whiting, M.F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogen. Evol. 2008, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ye, F.; Huang, Y. Mitochondrial genomes of four katydids (Orthoptera: Phaneropteridae): New gene rearrangements and their phylogenetic implications. Gene 2016, 575, 702–711. [Google Scholar] [CrossRef]
- Saenz Manchola, O.F.; Virrueta Herrera, S.; D’ Alessio, L.M.; Yoshizawa, K.; García Aldrete, A.N.; Johnson, K.P. Mitochondrial genomes within bark lice (Insecta: Psocodea: Psocomorpha) reveal novel gene rearrangements containing phylogenetic signal. Syst. Entomol. 2021, 46, 938–951. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics; Springer: Dordrecht, The Netherlands, 2000; pp. 133–147. [Google Scholar]
- Dowton, M.; Campbell, N.J. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Zhou, Z.; Guan, B.; Chai, J.; Che, X. Next-generation sequencing data used to determine the mitochondrial genomes and a preliminary phylogeny of Verophasmatodea insects. J. Asia-Pacif. Entomol. 2017, 20, 713–719. [Google Scholar] [CrossRef]
- Song, N.; Li, X.; Na, R. Mitochondrial genomes of stick insects (Phasmatodea) and phylogenetic considerations. PLoS ONE 2020, 15, e0240186. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Zhou, J.; Li, T.; Jiang, K.; Zhang, Y.; Zheng, C.; Liang, J.; Bu, W. The phylogenic position of aschiphasmatidae in euphasmatodea based on mitochondrial genomic evidence. Gene 2022, 808, 145974. [Google Scholar] [CrossRef]
- Günthe, K. Über die taxonomische Gliederung und die geographische Verbreitung der Insektenordnung der Phasmatodea. Beitr. Entomol. 1953, 3, 541–563. [Google Scholar]
- Bank, S.; Bradler, S. A second view on the evolution of flight in stick and leaf insects (Phasmatodea). BMC Ecol. Evo. 2022, 22, 62. [Google Scholar] [CrossRef]
- Simon, S.; Letsch, H.; Bank, S.; Buckley, T.R.; Donath, A.; Liu, S.; Machida, R.; Meusemann, K.; Misof, B.; Podsiadlowski, L. Old World and New World Phasmatodea: Phylogenomics resolve the evolutionary history of stick and leaf insects. Front. Ecol. Evol. 2019, 7, 345. [Google Scholar] [CrossRef]
- Tihelka, E.; Cai, C.; Giacomelli, M.; Pisani, D.; Donoghue, P.C. Integrated phylogenomic and fossil evidence of stick and leaf insects (Phasmatodea) reveal a Permian-Triassic co-origination with insectivores. R. Soc. Open Sci. 2020, 7, 201689. [Google Scholar] [CrossRef]
- Kômoto, N.; Yukuhiro, K.; Ueda, K.; Tomita, S. Exploring the molecular phylogeny of phasmids with whole mitochondrial genome sequences. Mol. Phylogen. Evol. 2011, 58, 43–52. [Google Scholar] [CrossRef]
- Büscher, T.H.; Buckley, T.R.; Grohmann, C.; Gorb, S.N.; Bradler, S. The evolution of tarsal adhesive microstructures in stick and leaf insects (Phasmatodea). Front. Ecol. Evol. 2018, 6, 69. [Google Scholar] [CrossRef]
- Bank, S.; Buckley, T.R.; Büscher, T.H.; Bresseel, J.; De Haan, M.; Dittmar, D.; Dräger, H.; Kahar, R.S.; Kang, A. Reconstructing the nonadaptive radiation of an ancient lineage of ground-dwelling stick insects (Phasmatodea: Heteropterygidae). Syst. Entomol. 2021, 46, 487–507. [Google Scholar] [CrossRef]
- Goldberg, J.; Bresseel, J.; Constant, J.; Kneubühler, B.; Leubner, F.; Michalik, P.; Bradler, S. Extreme convergence in egg-laying strategy across insect orders. Sci. Rep. 2015, 5, 7825. [Google Scholar] [CrossRef]
- Whiting, M.F.; Bradler, S.; Maxwell, T. Loss and recovery of wings in stick insects. Nature 2003, 421, 264–267. [Google Scholar] [CrossRef]
- Hennemann, F.H.; Conle, O.V.; Brock, P.D.; Seow Choen, F. Revision of the Oriental subfamily Heteropteryginae Kirby, 1896, with a rearrangement of the family Heteropterygidae and the descriptions of five new species of Haaniella Kirby, 1904. (Phasmatodea: Areolatae: Heteropterygidae). Zootaxa 2016, 4159, 1–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.K.; Chen, Q.P.; Ayivi, S.P.G.; Guan, J.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Three complete mitochondrial genomes of Orestes guangxiensis, Peruphasma schultei, and Phryganistria guangxiensis (Insecta: Phasmatodea) and their phylogeny. Insects 2021, 12, 779. [Google Scholar] [CrossRef] [PubMed]
- Glaw, F.; Hawlitschek, O.; Dunz, A.; Goldberg, J.; Bradler, S. When giant stick insects play with colors: Molecular phylogeny of the Achriopterini and description of two new splendid species (Phasmatodea: Achrioptera) from Madagascar. Front. Ecol. Evol. 2019, 7, 105. [Google Scholar] [CrossRef]
- Brock, P.D.; Hasenpusch, J. Studies on the leaf insects (Phasmida: Phylliidae) of Australia. J. Orthoptera Res. 2002, 11, 199–205. [Google Scholar] [CrossRef]
- Crampton, G. The lines of descent of the lower pterygotan insects, with notes on the relationships of the other forms. Entomol. News 1916, 27, 244–258. [Google Scholar]
- Friedemann, K.; Wipfler, B.; Bradler, S.; Beutel, R.G. On the head morphology of Phyllium and the phylogenetic relationships of Phasmatodea (Insecta). Acta Zool. 2012, 93, 184–199. [Google Scholar] [CrossRef]
- Tilgner, E.H. Systematics of Phasmida. Ph.D. Thesis, University of Georgia, Athens, GA, USA, 2002. [Google Scholar]
- Bank, S.; Cumming, R.T.; Li, Y.; Henze, K.; Le Tirant, S.; Bradler, S. A tree of leaves: Phylogeny and historical biogeography of the leaf insects (Phasmatodea: Phylliidae). Commun. Biol. 2021, 4, 932. [Google Scholar] [CrossRef]
- Forni, G.; Plazzi, F.; Cussigh, A.; Conle, O.; Hennemann, F.; Luchetti, A.; Mantovani, B. Phylomitogenomics provides new perspectives on the Euphasmatodea radiation (Insecta: Phasmatodea). Mol. Phylogen. Evol. 2021, 155, 106983. [Google Scholar] [CrossRef]
- Tomita, S.; Yukuhiro, K.; Kômoto, N. The mitochondrial genome of a stick insect Extatosoma tiaratum (Phasmatodea) and the phylogeny of polyneopteran insects. J. Insect Biotechnol. Sericol. 2011, 80, 3079–3088. [Google Scholar]
- Xu, K.K.; Chen, Q.P.; Guan, J.Y.; Zhang, Z.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. The mitochondrial genome of Eurycantha calcarata Lucas, 1869 (Phasmatodea: Lonchodinae) and its phylogeny. Mitochondrial DNA Part B 2021, 6, 3109–3111. [Google Scholar] [CrossRef]
- Bradley, J.C.; Galil, B. The Taxonomic Arrangement of the Phasmatodea with Keys to the Subfamilies and Tribes; Entomol. Soc. Wash: Washington, DC, USA, 1977; Volume 79, pp. 176–208. [Google Scholar]
- Bradler, S.; Robertson, J.A.; Whiting, M.F. A molecular phylogeny of Phasmatodea with emphasis on Necrosciinae, the most species-rich subfamily of stick insects. Syst. Entomol. 2014, 39, 205–222. [Google Scholar] [CrossRef]
- Sellick, J. The range of egg capsule morphology within the Phasmatodea and its relevance to the taxonomy of the order. Ital. J. Zool. 1997, 64, 97–104. [Google Scholar] [CrossRef]
- Zhang, L.P.; Yu, D.N.; Storey, K.B.; Cheng, H.Y.; Zhang, J.Y. Higher tRNA gene duplication in mitogenomes of praying mantises (Dictyoptera, Mantodea) and the phylogeny within Mantodea. Int. J. Biol. Macromol. 2018, 111, 787–795. [Google Scholar] [CrossRef]
- Lalitha, S. Primer premier 5. Biotech. Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; Volume 132, pp. 71–91. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogen. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Cameron, S. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst. Entomol. 2014, 39, 400–411. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Plazzi, F.; Ricci, A.; Passamonti, M. The mitochondrial genome of Bacillus stick insects (Phasmatodea) and the phylogeny of orthopteroid insects. Mol. Phylogen. Evol. 2011, 58, 304–316. [Google Scholar] [CrossRef]
- Cameron, S.L.; Barker, S.C.; Whiting, M.F. Mitochondrial genomics and the new insect order Mantophasmatodea. Mol. Phylogen. Evol. 2006, 38, 274–279. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- FigTree Version 1.4.0. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 March 2023).
- Xia, Y.; Zheng, Y.; Murphy, R.W.; Zeng, X. Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genom. 2016, 17, 965. [Google Scholar] [CrossRef]
- Lunt, D.H.; Whipple, L.E.; Hyman, B.C. Mitochondrial DNA variable number tandem repeats (VNTRs): Utility and problems in molecular ecology. Mol. Ecol. 1998, 7, 1441–1455. [Google Scholar] [CrossRef]
- Wang, X.; Liu, N.; Zhang, H.; Yang, X.J.; Huang, Y.; Lei, F. Extreme variation in patterns of tandem repeats in mitochondrial control region of yellow-browed tits (Sylviparus modestus, Paridae). Sci. Rep. 2015, 5, 13227. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Solignac, M.; Monerot, M.; Mounolou, J.C. Concerted evolution of sequence repeats in Drosophila mitochondrial DNA. J. Mol. Evol. 1986, 24, 53–60. [Google Scholar] [CrossRef]
- Dowton, M.; Castro, L.; Austin, A. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome ‘morphology’. Invertebr. Syst. 2002, 16, 345–356. [Google Scholar] [CrossRef]
- Guo, J.; Yan, Z.T.; Fu, W.B.; Yuan, H.; Li, X.D.; Chen, B. Complete mitogenomes of Anopheles peditaeniatus and Anopheles nitidus and phylogenetic relationships within the genus Anopheles inferred from mitogenomes. Parasites Vectors 2021, 14, 452. [Google Scholar] [CrossRef]
- Dowton, M.; Austin, A.D. Increased genetic diversity in mitochondrial genes is correlated with the evolution of parasitism in the Hymenoptera. J. Mol. Evol. 1995, 41, 958–965. [Google Scholar] [CrossRef]
- Timmermans, M.J.; Vogler, A.P. Phylogenetically informative rearrangements in mitochondrial genomes of Coleoptera, and monophyly of aquatic elateriform beetles (Dryopoidea). Mol. Phylogen. Evol. 2012, 63, 299–304. [Google Scholar] [CrossRef]
- Shao, R.; Barker, S.C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 2003, 20, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, P.Y.; Xue, X.F.; Hua, H.Q.; Li, Y.X.; Zhang, F.; Wei, S.J. Extensive gene rearrangements in the mitochondrial genomes of two egg parasitoids, Trichogramma japonicum and Trichogramma ostriniae (Hymenoptera: Chalcidoidea: Trichogrammatidae). Sci. Rep. 2018, 8, 7034. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, H.; Feng, Z.; Li, B.; Zhou, W.; Song, F.; Li, H.; Zhang, L.; Cai, W. Novel gene rearrangement in the mitochondrial genome of Pachyneuron aphidis (Hymenoptera: Pteromalidae). Int. J. Biol. Macromol. 2020, 149, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wu, Y.; Yang, C.; Gu, X.; Wilson, J.J.; Li, H.; Cai, W.; Yang, H.; Song, F. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int. J. Biol. Macromol. 2020, 164, 540–547. [Google Scholar] [CrossRef]
- Dowton, M.; Castro, L.R.; Campbell, S.L.; Bargon, S.D.; Austin, A.D. Frequent mitochondrial gene rearrangements at the hymenopteran nad3–nad5 junction. J. Mol. Evol. 2003, 56, 517–526. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Sharkey, M.J.; van Achterberg, C.; Chen, X.X. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genom. 2010, 11, 371. [Google Scholar] [CrossRef]
- Mueller, R.L.; Boore, J.L. Molecular mechanisms of extensive mitochondrial gene rearrangement in plethodontid salamanders. Mol. Biol. Evol. 2005, 22, 2104–2112. [Google Scholar] [CrossRef]
- Boore, J.L.; Collins, T.M.; Stanton, D.; Daehler, L.L.; Brown, W.M. Deducing the pattern of arthropod phytogeny from mitochondrial DNA rearrangements. Nature 1995, 376, 163–165. [Google Scholar] [CrossRef]
- Li, Y.; Wang, S.; Chen, J.; Zhou, J.; Bu, W. Two new stick insect species of Sosibia Stål (Phasmatodea: Lonchodidae: Necrosciinae) from China and the first report on mitochondrial genomes of this genus. Arch. Insect Biochem. Physiol. 2022, 111, e21901. [Google Scholar] [CrossRef]
- Zompro, O. Abhandlungen des Naturwissenschftlichen Vereins in Hambur. In Revision of the Genera of the Areolatae, Including the Status of Timema and Agathemera (Insecta, Phasmatodea); Goecke & Evers: Baden-Wuerttemberg, Germany, 2004; Volume 37, pp. 1–327. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, X. Organization of the mitochondrial genome of Ramulus irregulatiter dentatus (Phasmatidae: Phasmatidae). Front. Genet. 2022, 13, 967113. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Sequence Alignments | ||
|---|---|---|
| Subset | Subset Partitions | Best Model |
| Partition 1 | ND6_codon1, ATP6_codon1, ND3_codon1, ND2_codon1, ATP8_codon1 | GTR + I + G |
| Partition 2 | Cytb_codon2, COX3_codon2, COX2_codon2, ND3_codon2, ND2_codon2, ATP6_codon2 | GTR + I + G |
| Partition 3 | ND2_codon3, ATP6_codon3, Cytb_codon3, ND6_codon3, ND3_codon3, ATP8_codon3 | GTR + G |
| Partition 4 | ATP8_codon2, ND2_codon2, ND6_codon2 | GTR + I + G |
| Partition 5 | COX1_codon1 | GTR + I + G |
| Partition 6 | COX1_codon2 | TVM + I + G |
| Partition 7 | COX1_codon3 | GTR + I + G |
| Partition 8 | Cytb_codon1, COX3_codon1, COX2_codon1 | GTR + I + G |
| Partition 9 | COX3_codon3, COX2_codon3 | GTR + I + G |
| Partition 10 | ND1_codon1, ND5_codon1 | TRN + I + G |
| Partition 11 | ND4L_codon2, ND5_codon2, ND1_codon2 | GTR + G |
| Partition 12 | ND4L_codon3, ND1_codon3, ND4_codon3, ND5_codon3 | TVM + I + G |
| Partition 13 | ND4_codon1, ND4L_codon1 | TVM + I + G |
| Partition 14 | ND4_codon2 | GTR + I + G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Zhang, L.; Li, K.; Hong, Y.; Storey, K.B.; Zhang, J.; Yu, D. Nine Mitochondrial Genomes of Phasmatodea with Two Novel Mitochondrial Gene Rearrangements and Phylogeny. Insects 2023, 14, 485. https://doi.org/10.3390/insects14050485
Yuan Y, Zhang L, Li K, Hong Y, Storey KB, Zhang J, Yu D. Nine Mitochondrial Genomes of Phasmatodea with Two Novel Mitochondrial Gene Rearrangements and Phylogeny. Insects. 2023; 14(5):485. https://doi.org/10.3390/insects14050485
Chicago/Turabian StyleYuan, Yani, Lihua Zhang, Ke Li, Yuehuan Hong, Kenneth B. Storey, Jiayong Zhang, and Danna Yu. 2023. "Nine Mitochondrial Genomes of Phasmatodea with Two Novel Mitochondrial Gene Rearrangements and Phylogeny" Insects 14, no. 5: 485. https://doi.org/10.3390/insects14050485