
Comparative Mitogenomics of Jumping Spiders with First Complete Mitochondrial Genomes of Euophryini (Araneae: Salticidae)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. DNA Extraction, Library Preparation and Sequencing
2.3. Mitochondrial Genome Assembly, Annotation and Sequence Analysis
2.4. Phylogenetic Analyses
3. Results
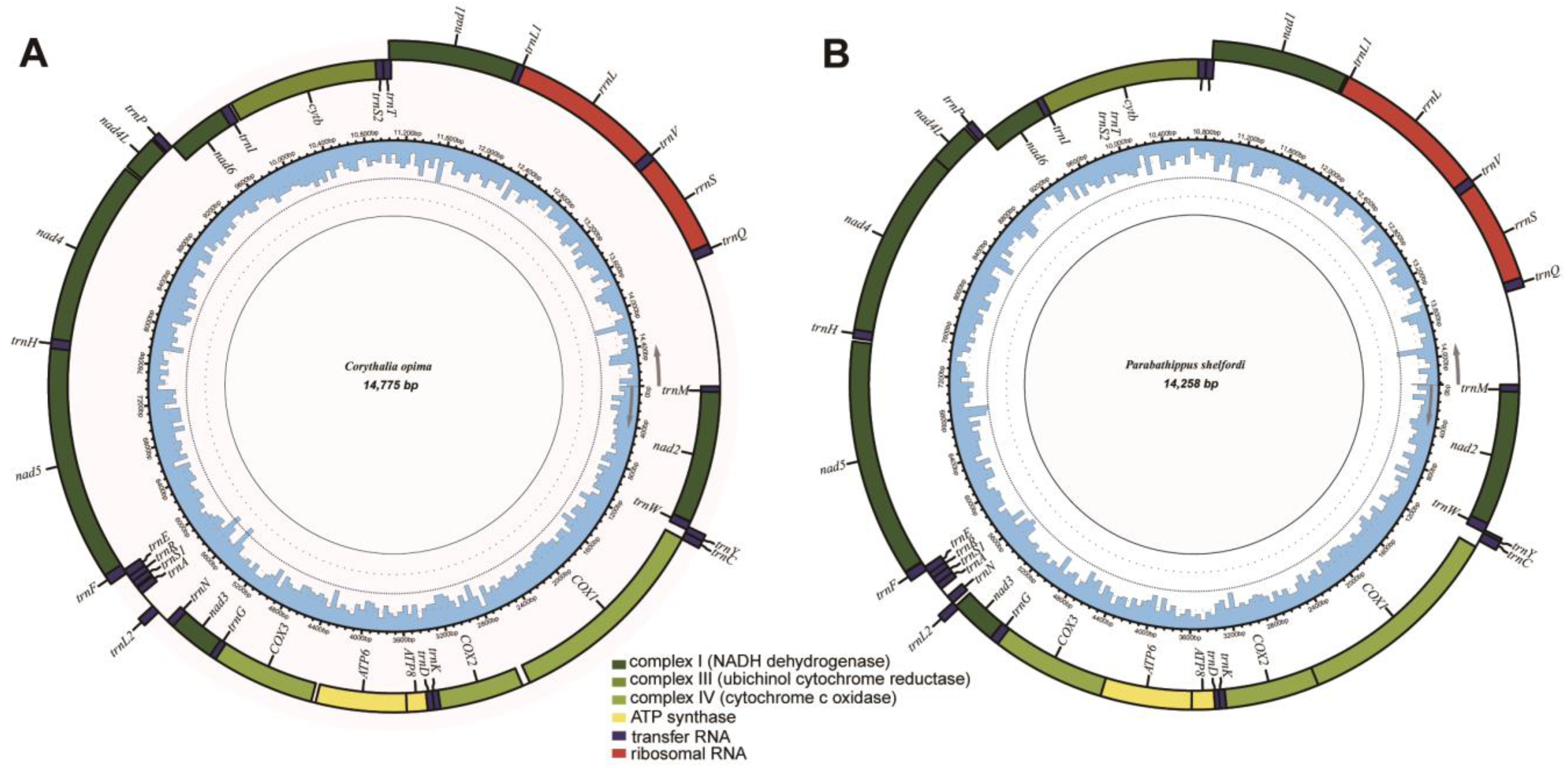
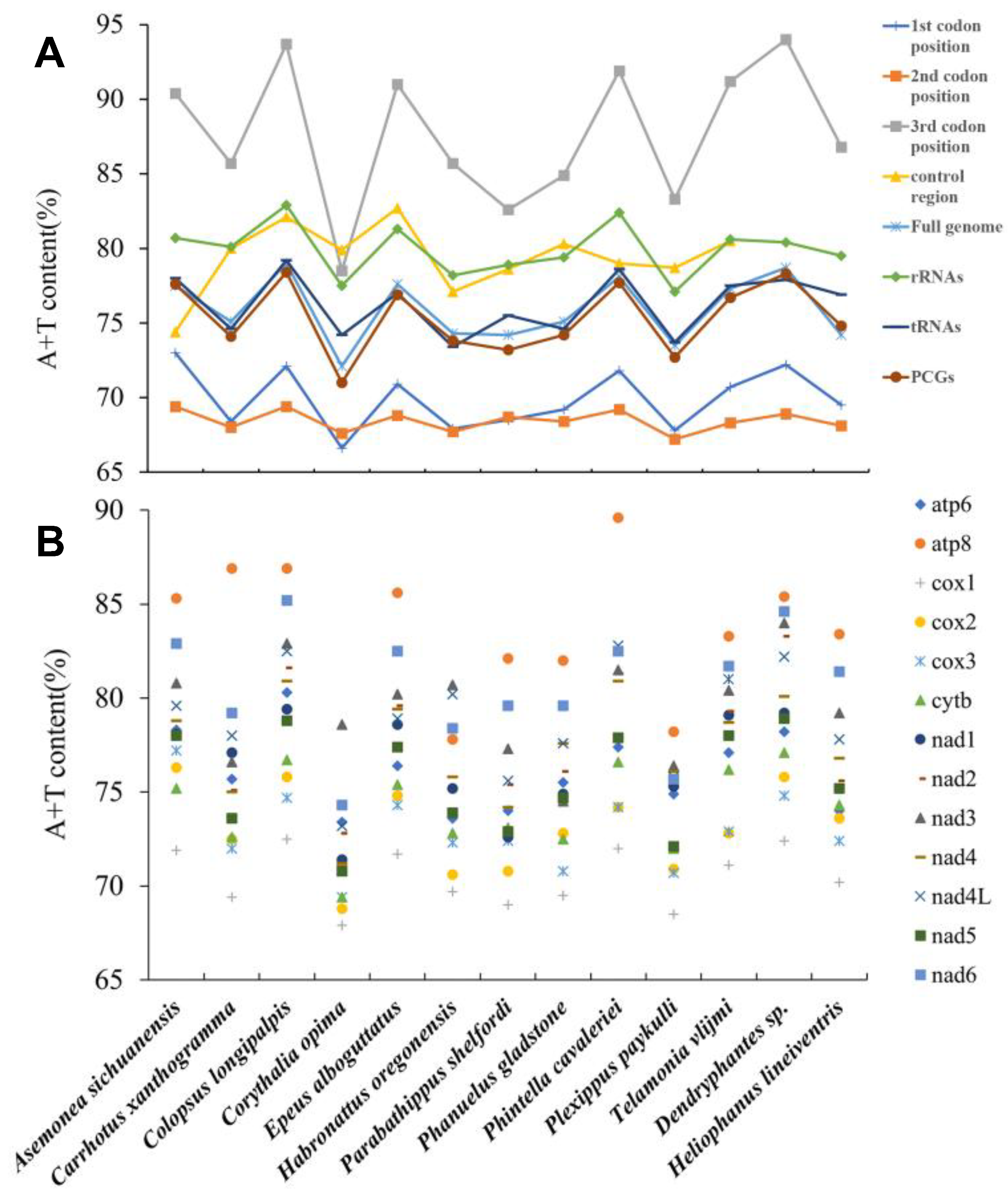
3.1. General Features of Mitogenomes of Euophryini
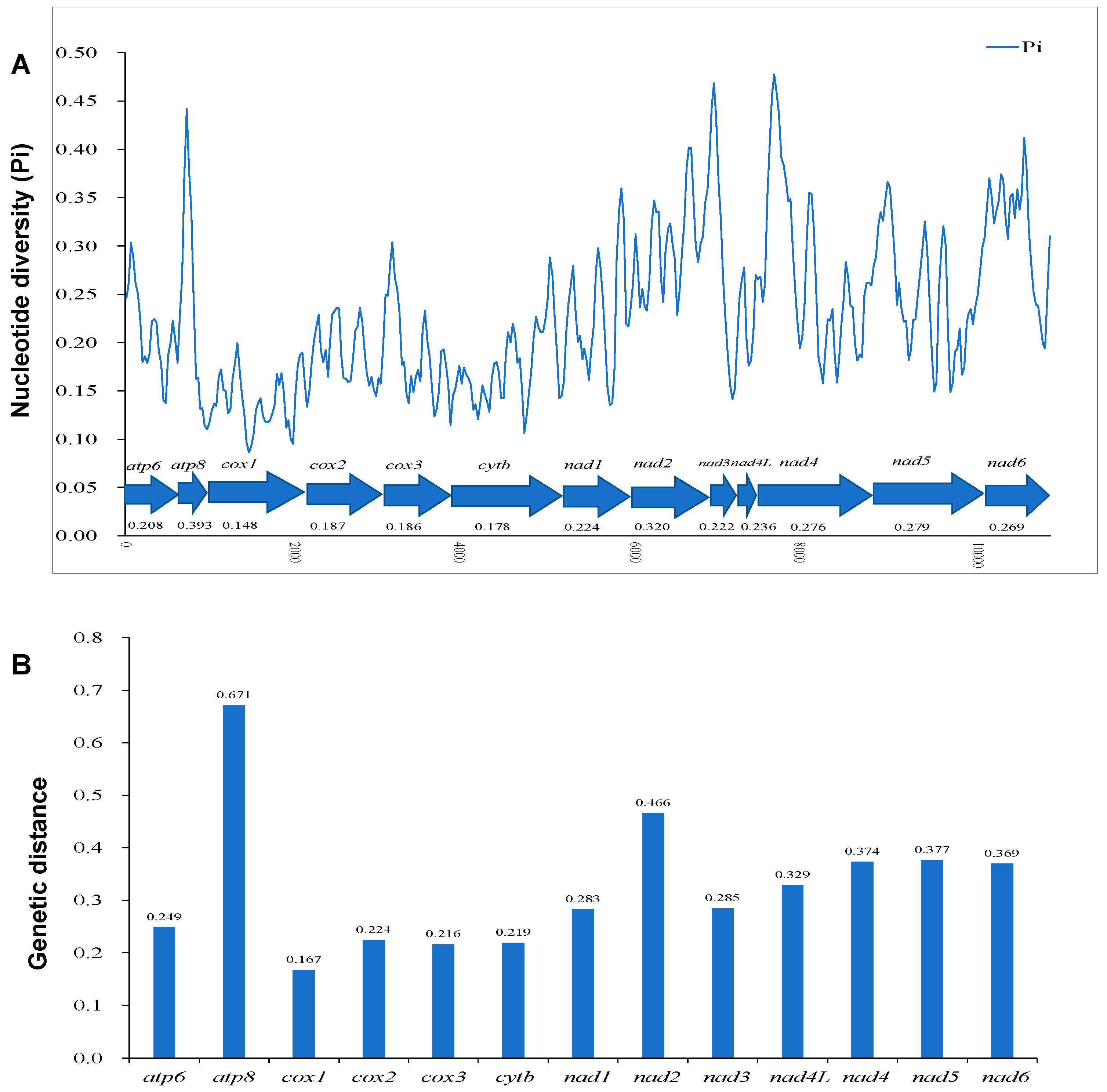
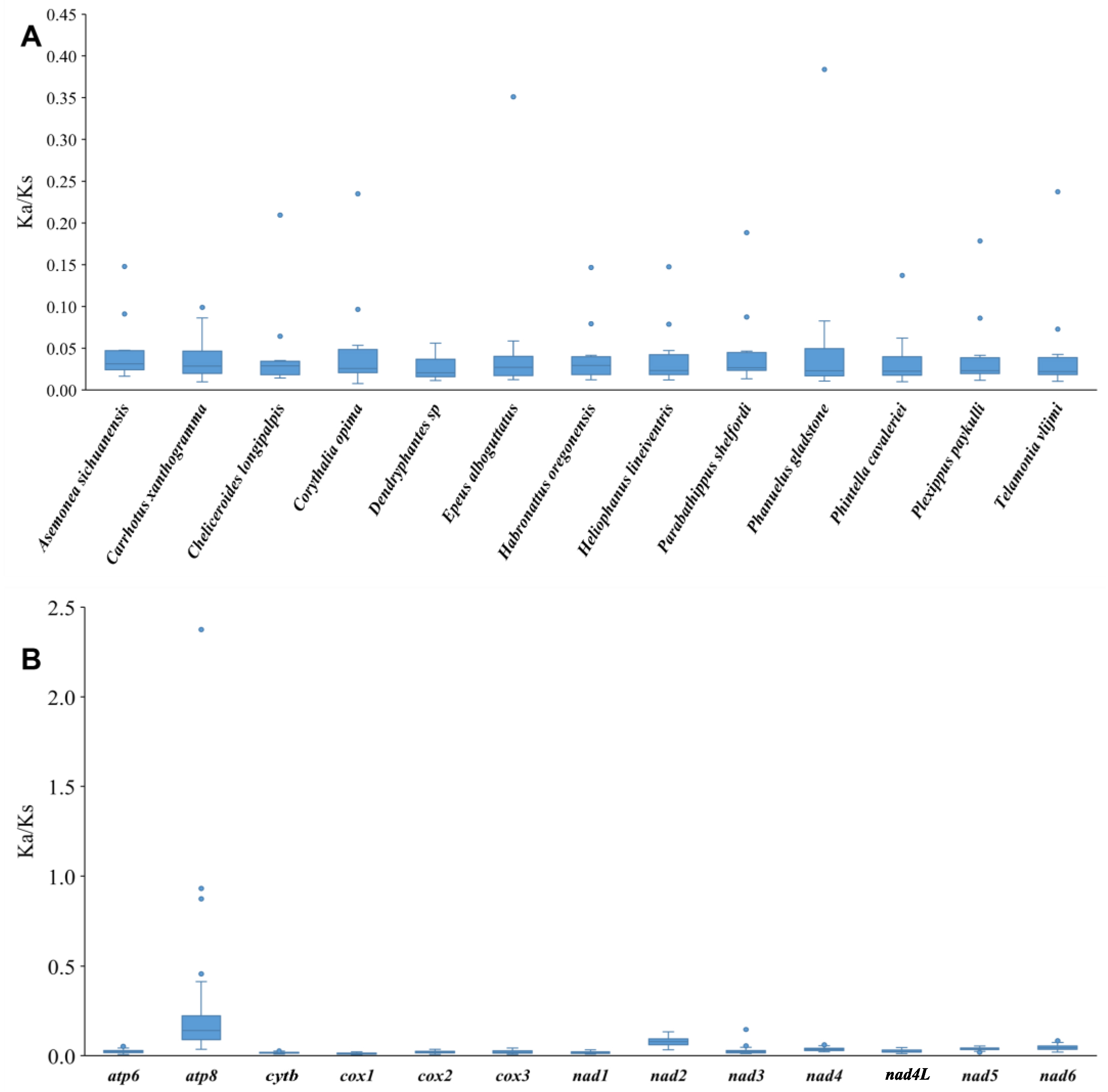
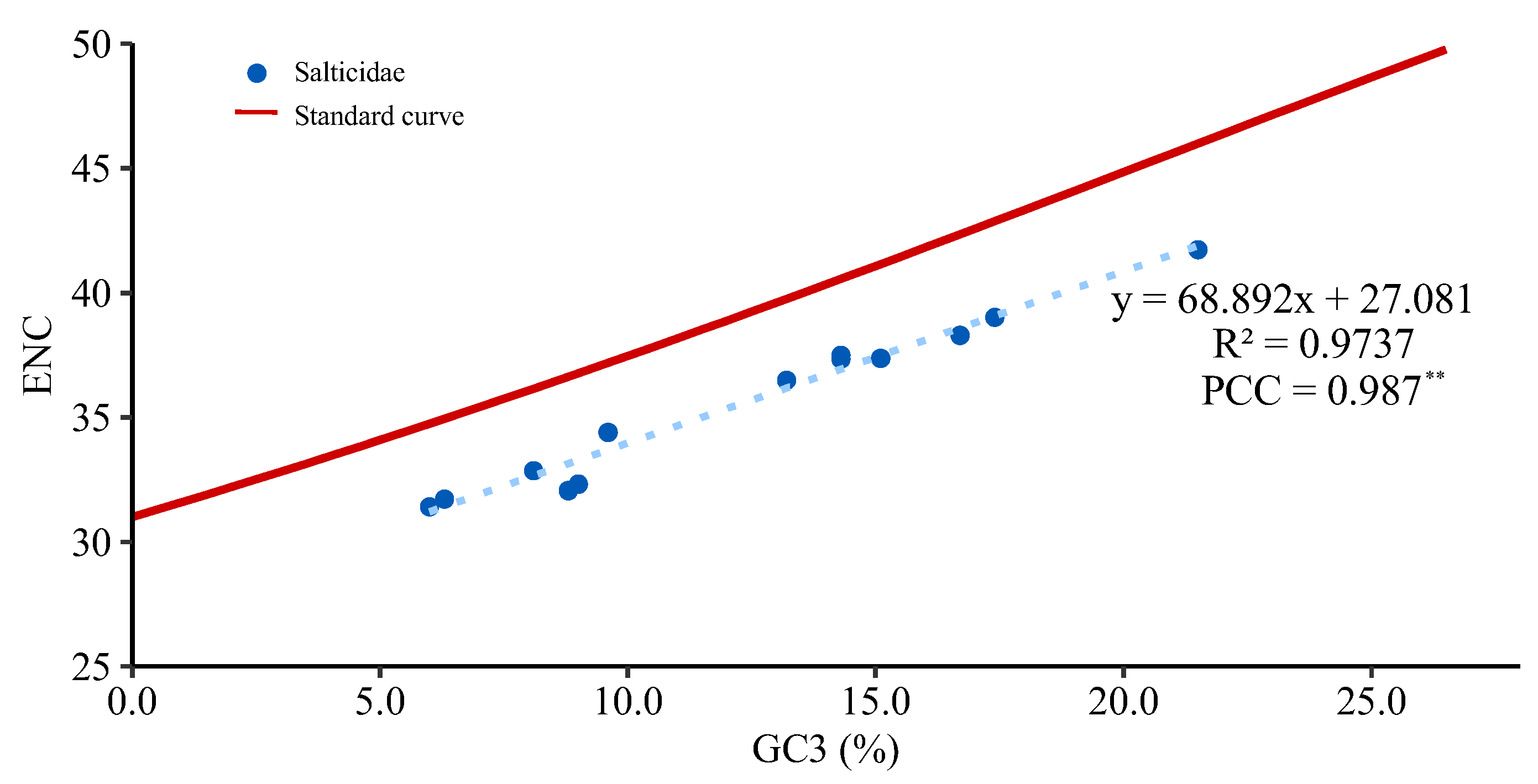
3.2. Protein-Coding Genes
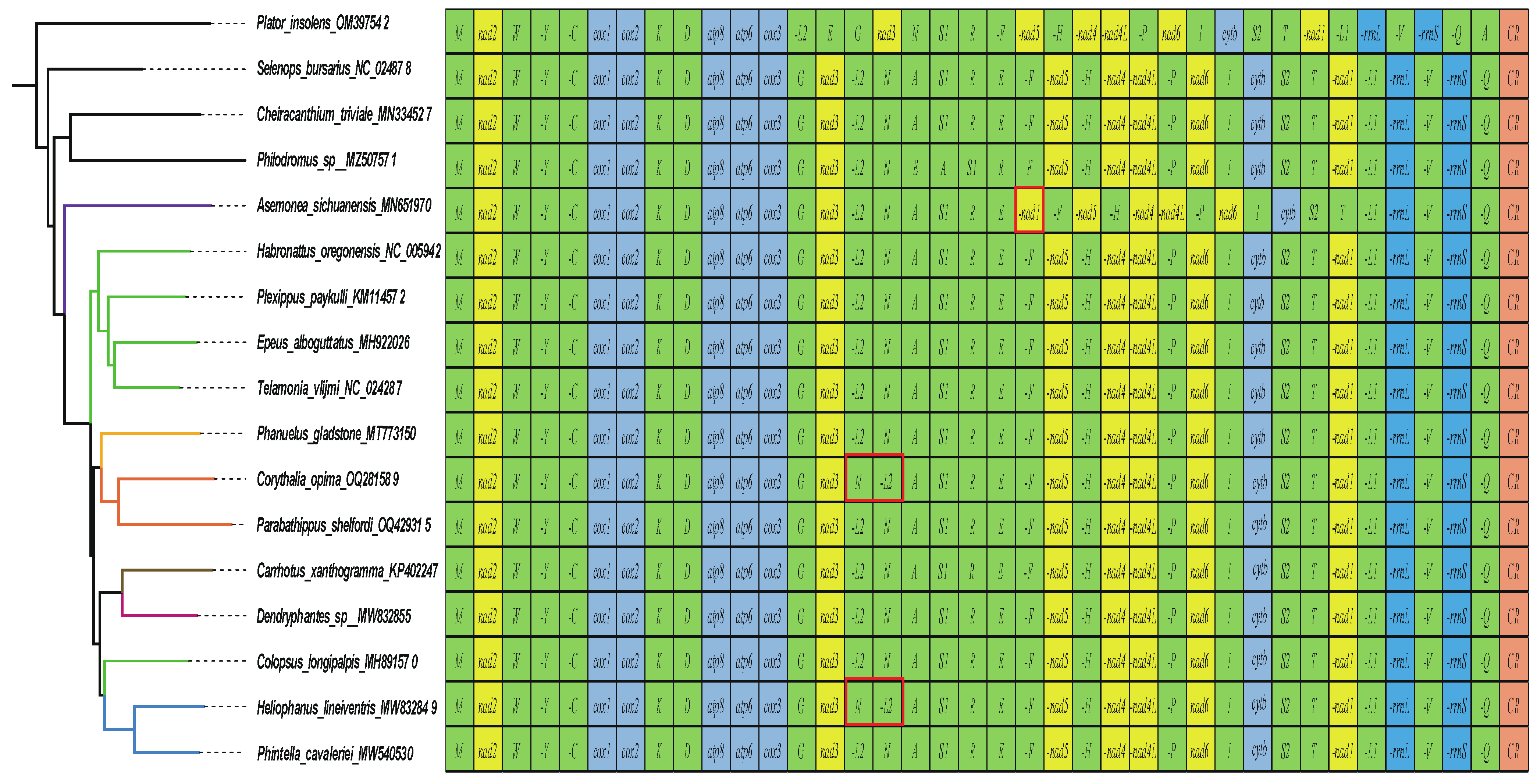
3.3. Transfer and Ribosomal RNA Genes
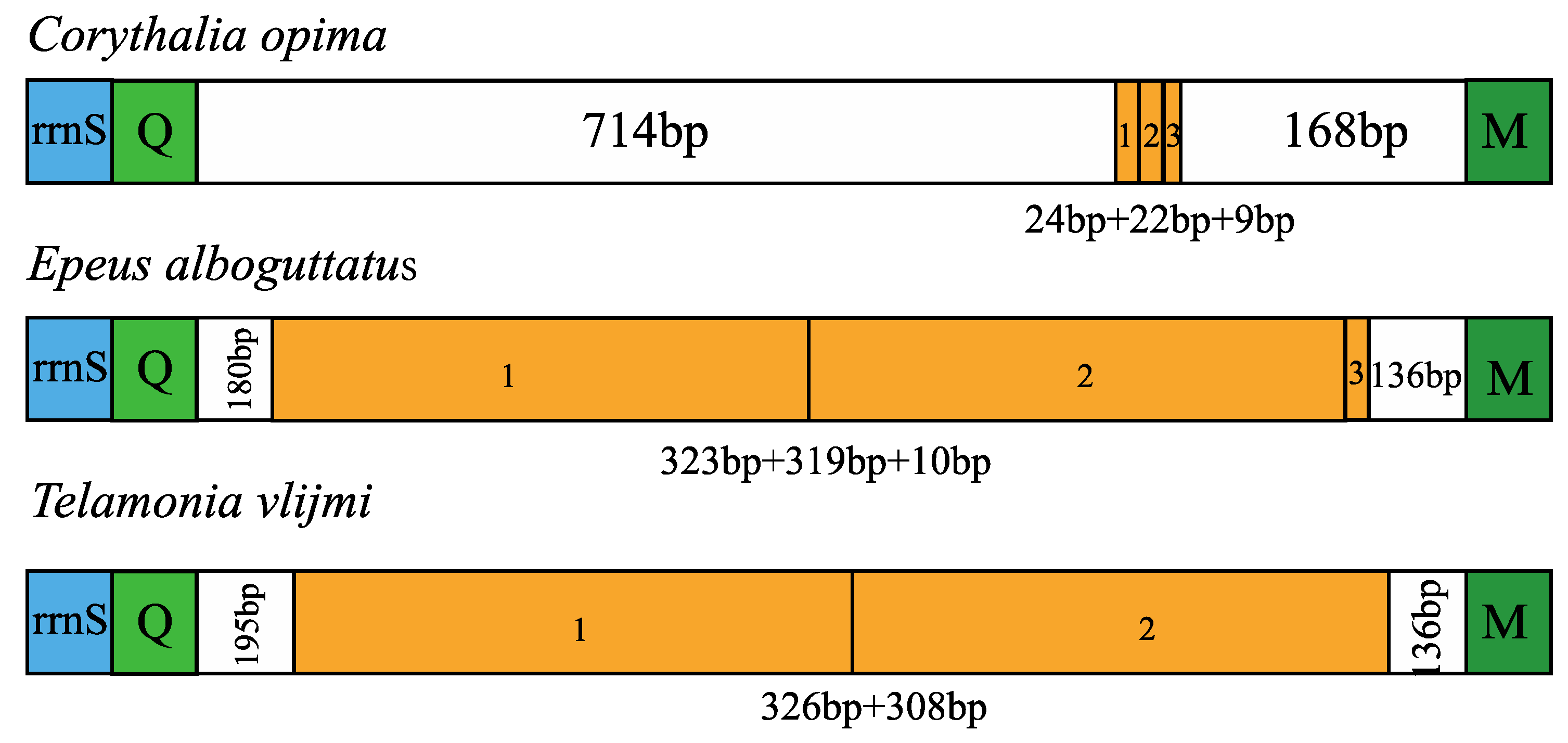
3.4. Control Region
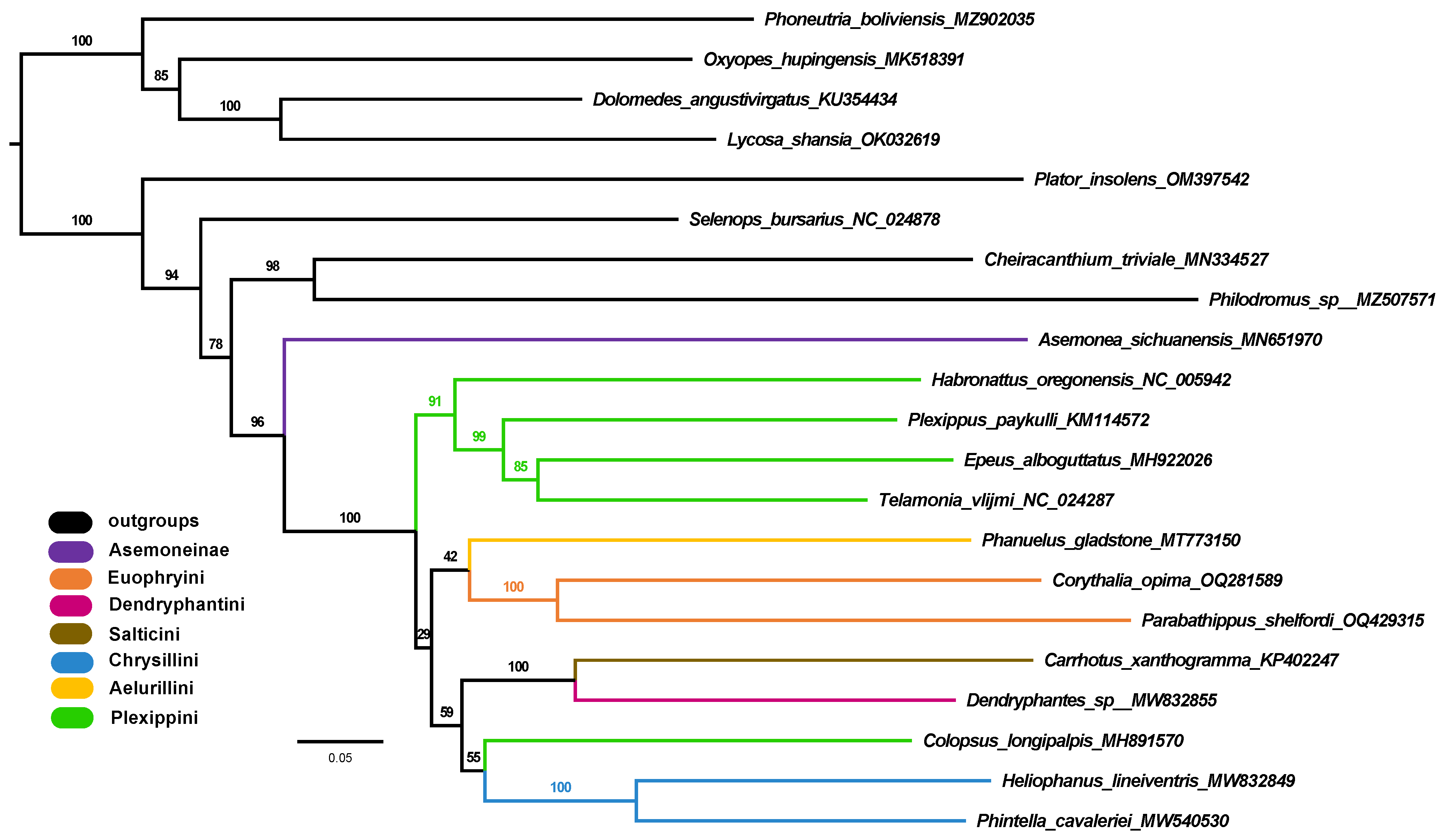
3.5. Phylogenetic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L.; Collins, T.M.; Stanton, D.; Daehler, L.L.; Brown, W.M. Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature 1995, 376, 163–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary patterns of the mitochondrial genome in Metazoa: Exploring the role of mutation and selection in mitochondrial protein-coding genes. Genome Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Kern, E.M.A.; Kim, T.; Park, J.K. The mitochondrial genome in nematode phylogenetics. Front. Ecol. Evol. 2020, 8, 250. [Google Scholar] [CrossRef]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Liu, T.; Kang, Y.; Liu, H.Y.; Yang, Y.X. First complete mitochondrial genomes of Ototretinae (Coleoptera, Lampyridae) with evolutionary insights into the gene rearrangement. Genomics 2022, 114, 110305. [Google Scholar] [CrossRef]
- Arribas, P.; Andújar, C.; Moraza, M.L.; Linard, B.; Emerson, B.C.; Vogler, A.P. Mitochondrial metagenomics reveals the ancient origin and phylodiversity of soil mites and provides a phylogeny of the Acari. Mol. Biol. Evol. 2020, 37, 683–694. [Google Scholar] [CrossRef]
- Lopardo, L.; Uhl, G. Testing mitochondrial marker efficacy for DNA barcoding in spiders: A test case using the dwarf spider genus Oedothorax (Araneae: Linyphiidae: Erigoninae). Invertebr. Syst. 2014, 28, 501–521. [Google Scholar] [CrossRef]
- Xu, C.C.Y.; Yen, I.J.; Bowman, D.; Turner, C.R. Spider web DNA: A new spin on noninvasive genetics of predator and prey. PLoS ONE 2015, 10, e0142503. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuwenhuyse, P.; Demaeght, P.; Dermauw, W.; Khalighi, M.; Stevens, C.V.; Vanholme, B.; Tirry, L.; Lümmen, P.; Van Leeuwen, T. On the mode of action of bifenazate: New evidence for a mitochondrial target site. Pestic. Biochem. Physiol. 2012, 104, 88–95. [Google Scholar] [CrossRef]
- Pichaud, N.; Ballard, J.W.O.; Tanguay, R.M.; Blier, P.U. Naturally occurring mitochondrial DNA haplotypes exhibit metabolic differences: Insight into functional properties of mitochondria. Evolution 2012, 66, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Masta, S.E.; Boore, J.L. Parallel evolution of truncated transfer RNA genes in arachnid mitochondrial genomes. Mol. Biol. Evol. 2008, 25, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Pons, J.; Bover, P.; Bidegaray-Batista, L.; Arnedo, M.A. Arm-less mitochondrial tRNAs conserved for over 30 millions of years in spiders. BMC Genom. 2019, 20, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Lv, Y.; Wen, Z.; Bian, C.; Zhang, X.; Guo, S.; Shi, Q.; Li, D. The complete mitochondrial genome of the intertidal spider (Desis jiaxiangi) provides novel insights into the adaptive evolution of the mitogenome and the evolution of spiders. BMC Ecol. Evol. 2021, 21, 72. [Google Scholar] [CrossRef]
- Tyagi, K.; Kumar, V.; Poddar, N.; Prasad, P.; Tyagi, I.; Kundu, S.; Chandra, K. The gene arrangement and phylogeny using mitochondrial genomes in spiders (Arachnida: Araneae). Int. J. Biol. Macromol. 2020, 146, 488–496. [Google Scholar] [CrossRef]
- Li, M.; Chen, W.T.; Zhang, Q.L.; Liu, M.; Xing, C.W.; Cao, Y.; Luo, F.Z.; Yuan, M.L. Mitochondrial phylogenomics provides insights into the phylogeny and evolution of spiders (Arthropoda: Araneae). Zool. Res. 2022, 43, 566–584. [Google Scholar] [CrossRef]
- Platnick, N.I. The World Spider Catalog, Natural History Museum Bern, online at version 24.0. 2023. Available online: https://wsc.nmbe.ch (accessed on 1 March 2023).
- Maddison, W.P. A phylogenetic classification of jumping spiders (Araneae: Salticidae). J. Arachnol. 2015, 43, 231–292. [Google Scholar] [CrossRef]
- Xia, P.L.; Huang, Y.; Xie, X.F.; Yan, S.Y.; Luo, Y.; Yang, W.J. The complete mitochondrial genome and phylogenetic analysis of Phintella cavaleriei (Araneae: Salticidae). Mitochondrial DNA Part B Resour. 2021, 6, 1625–1626. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yan, X.; Xu, K.; Yang, W.; Li, C. The complete mitochondrial genome of Epeus alboguttatus (Araneae: Salticidae). Mitochondrial DNA Part B Resour. 2019, 4, 316–317. [Google Scholar] [CrossRef] [Green Version]
- Masta, S.E.; Boore, J.L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 2004, 21, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Wang, J.; Zhao, X.; Liu, H.; Zhu, S. Mitochondrial genomes of two Lycosa spiders (Araneae, Lycosidae): Genome description and phylogenetic implications. Diversity 2022, 14, 538. [Google Scholar] [CrossRef]
- Yang, W.J.; Xu, K.K.; Yang, D.X.; Li, C. The complete mitochondrial genome of Oxyopes hupingensis (Araneae: Oxyopidae): Characterization and phylogenetic analysis. Mitochondrial DNA Part B Resour. 2019, 4, 1780–1781. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-L.; Huang, J.; Li, M.-Y.; Yu, X.-P. The complete mitochondrial genome of a nursery-web spider Dolomedes angustivirgatus (Araneae: Pisauridae). Mitochondrial DNA Part B 2020, 5, 1695–1696. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xu, K.; Yan, Y.; Yang, W.; Yang, H.; Li, C.; Yang, D. The complete mitochondrial genome of a jumping spider, Cheliceroides longipalpis Zabka (Araneae: Salticidae). Mitochondrial DNA Part B Resour. 2019, 4, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.J.; Fang, H.Y.; Zhang, P.; Pan, H.C. The complete mitochondrial genome of pantropical jumping spider Plexippus paykulli (Araneae: Salticidae). Mitochondrial DNA 2016, 27, 1490–1491. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yoon, K.B.; Park, Y.C. The complete mitochondrial genome of the jumping spider Telamonia vlijmi (Araneae: Salticidae). Mitochondrial DNA 2016, 27, 635–636. [Google Scholar] [CrossRef]
- Fang, W.Y.; Wang, Z.L.; Li, C.; Yang, X.Q.; Yu, X.P. The complete mitogenome of a jumping spider Carrhotus xanthogramma (Araneae: Salticidae) and comparative analysis in four salticid mitogenomes. Genetica 2016, 144, 699–709. [Google Scholar] [CrossRef]
- Pan, W.J.; Fang, H.Y.; Zhang, P.; Pan, H.C. The complete mitochondrial genome of flat spider Selenops bursarius (Araneae: Selenopidae). Mitochondrial DNA 2016, 27, 1488–1489. [Google Scholar] [CrossRef]
- Ding, Y.; Xu, H.; Ma, H.; Zhang, J. The complete mitogenome of Plator insolens (Araneae: Trochanteriidae) with phylogenetic implication. Mitochondrial DNA Part B Resour. 2022, 7, 1032–1034. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Pütz, J.; Florentz, C.; Donath, A.; Jühling, F.; Stadler, P.F.; Externbrink, F.; Fritzsch, G.; Middendorf, M. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating selective pressure on coding and non-coding sequences. Genom. Proteom. Bioinf. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef]
- Wei, S.J.; Shi, M.; Chen, X.X.; Sharkey, M.J.; van Achterberg, C.; Ye, G.Y.; He, J.H. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 2010, 5, e12708. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Xu, K.K.; Yang, D.X.; Li, C.; Yang, W.J. Characterization of the complete mitochondrial genome and phylogenetic analysis of Leucauge celebesiana (Araneae: Tetragnathidae). Mitochondrial DNA Part B Resour. 2019, 4, 3314–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.J.; Xu, K.K.; Liu, Y.; Yang, D.X.; Li, C. Complete mitochondrial genome and phylogenetic analysis of Argiope perforata (Araneae: Araneidae). Mitochondrial DNA Part B Resour. 2019, 4, 1963–1964. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Xu, K.K.; Yang, D.X.; Li, C.; Yang, W.J. The complete mitochondrial genome of Argiope ocula (Araneae: Araneidae) and its phylogeny. Mitochondrial DNA Part B Resour. 2019, 4, 3318–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Tyagi, K.; Chakraborty, R.; Prasad, P.; Kundu, S.; Tyagi, I.; Chandra, K. The complete mitochondrial genome of endemic giant tarantula, Lyrognathus crotalus (Araneae: Theraphosidae) and comparative analysis. Sci. Rep. 2020, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Song, D.; Zhou, K.; Sun, H. The mitochondrial sequences of Heptathela hangzhouensis and Ornithoctonus huwena reveal unique gene arrangements and atypical tRNAs. J. Mol. Evol. 2005, 60, 57–71. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Miura, S.; Yamada, C.; Hashiguchi, Y. Gene rearrangements in gekkonid mitochondrial genomes with shuffling, loss, and reassignment of tRNA genes. BMC Genomics 2014, 15, 930. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Miao, X.G.; Kong, X.Y. A novel model of double replications and random loss accounts for rearrangements in the mitogenome of Samariscus latus (Teleostei: Pleuronectiformes). BMC Genomics 2014, 15, 352. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA modifications: Impact on structure and thermal adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity (Edinb). 2008, 101, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Dávila, S.; Piñero, D.; Bustos, P.; Cevallos, M.A.; Dávila, G. The mitochondrial genome sequence of the scorpion Centruroides limpidus (Karsch 1879) (Chelicerata; Arachnida). Gene 2005, 360, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.F.; Deng, W.; Qu, S.X.; Hong, X.Y.; Shao, R. The mitochondrial genomes of sarcoptiform mites: Are any transfer RNA genes really lost? BMC Genomics 2018, 19, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giegé, R.; Jühling, F.; Pütz, J.; Stadler, P.; Sauter, C.; Florentz, C. Structure of transfer RNAs: Similarity and variability. Wiley Interdiscip. Rev. RNA 2012, 3, 37–61. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef]
- Zhang, P.; Fang, H.Y.; Pan, W.J.; Pan, H.C. The complete mitochondrial genome of the writing spider Argiope amoena (Araneae: Araneidae). Mitochondrial DNA 2016, 27, 1492–1493. [Google Scholar] [CrossRef]
- Zhang, P.; Fang, H.Y.; Pan, W.J.; Pan, H.C. The complete mitochondrial genome of the wasp spider Argiope bruennichi (Araneae: Araneidae). Mitochondrial DNA 2016, 27, 996–997. [Google Scholar] [CrossRef]
- Pan, W.J.; Fang, H.Y.; Zhang, P.; Pan, H.C. The complete mitochondrial genome of Nephila clavata (Araneae: Nephilidae) Chinese population. Mitochondrial DNA 2016, 27, 994–995. [Google Scholar] [CrossRef]
- Hu, M.; Jex, A.R.; Campbell, B.E.; Gasser, R.B. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat. Protoc. 2007, 2, 2339–2344. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Z.; Peng, Z. The mitochondrial genome of the water spider Argyroneta aquatica (Araneae: Cybaeidae). Zool. Scr. 2015, 44, 179–190. [Google Scholar] [CrossRef]
- Sun, J.T.; Jin, P.Y.; Hoffmann, A.A.; Duan, X.Z.; Dai, J.; Hu, G.; Xue, X.F.; Hong, X.Y. Evolutionary divergence of mitochondrial genomes in two Tetranychus species distributed across different climates. Insect Mol. Biol. 2018, 27, 698–709. [Google Scholar] [CrossRef]
- Chang, H.; Qiu, Z.; Yuan, H.; Wang, X.; Li, X.; Sun, H.; Guo, X.; Lu, Y.; Feng, X.; Majid, M.; et al. Evolutionary rates of and selective constraints on the mitochondrial genomes of Orthoptera insects with different wing types. Mol. Phylogenet. Evol. 2020, 145, 106734. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.Y.; Liang, L.; Zhu, Z.H.; Zhou, W.P.; Irwin, D.M.; Zhang, Y.P. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Meng, T.; Wei, W. Analysis of synonymous codon usage bias in helicase gene from Autographa californica multiple nucleopolyhedrovirus. Genes Genom. 2018, 40, 767–780. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, X. Codon usage bias and its influencing factors in the chloroplast genome of Macadamia integrifolia Maiden & Betche. Plant Sci. J. 2022, 40, 229–239. [Google Scholar]
- Fernández, R.; Kallal, R.J.; Dimitrov, D.; Ballesteros, J.A.; Arnedo, M.A.; Giribet, G.; Hormiga, G. Phylogenomics, diversification dynamics, and comparative transcriptomics across the spider tree of life. Curr. Biol. 2018, 28, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Garrison, N.L.; Rodriguez, J.; Agnarsson, I.; Coddington, J.A.; Griswold, C.E.; Hamilton, C.A.; Hedin, M.; Kocot, K.M.; Ledford, J.M.; Bond, J.E. Spider phylogenomics: Untangling the spider tree of life. PeerJ 2016, 4, e1719. [Google Scholar] [CrossRef] [Green Version]
- Kallal, R.J.; Kulkarni, S.S.; Dimitrov, D.; Benavides, L.R.; Arnedo, M.A.; Giribet, G.; Hormiga, G. Converging on the orb: Denser taxon sampling elucidates spider phylogeny and new analytical methods support repeated evolution of the orb web. Cladistics 2021, 37, 298–316. [Google Scholar] [CrossRef]
- Azevedo, G.H.F.; Bougie, T.; Carboni, M.; Hedin, M.; Ramírez, M.J. Combining genomic, phenotypic and Sanger sequencing data to elucidate the phylogeny of the two-clawed spiders (Dionycha). Mol. Phylogenet. Evol. 2022, 166, 107327. [Google Scholar] [CrossRef]
- Maddison, W.P.; Evans, S.C.; Hamilton, C.A.; Bond, J.E.; Lemmon, A.R.; Lemmon, E.M. A genome-wide phylogeny of jumping spiders (Araneae, Salticidae), using anchored hybrid enrichment. ZooKeys 2017, 695, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Żabka, M. Systematic and zoogeographic study on the family Salticidae (Araneae) from Viet-Nam. Annales Zoologici, Warszawa 1985, 39, 197–485. [Google Scholar]
- Logunov, D.V. Jumping spiders (Araneae: Salticidae) of the Na Hang Nature Reserve, Tuyen Quang Province, Vietnam. Arachnology 2021, 18, 1021–1055. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Tribe | Species | GenBank Accession Number | Reference |
|---|---|---|---|---|---|
| Cheiracanthiidae | Cheiracanthium triviale | MN334527 | [16] | ||
| Ctenidae | Phoneutria boliviensis | MZ902035 | Unpublished | ||
| Lycosidae | Lycosa shansia | OK032619 | [23] | ||
| Oxyopidae | Oxyopes hupingensis | MK518391 | [24] | ||
| Philodromidae | Philodromus sp. | MZ507571 | Unpublished | ||
| Pisauridae | Dolomedes angustivirgatus | KU354434 | [25] | ||
| Salticidae | Asemoneinae | Asemonea sichuanensis | MN651970 | Unpublished | |
| Salticinae | Aelurillini | Phanuelus gladstone | MT773150 | Unpublished | |
| Chrysillini | Heliophanus lineiventris | MW832849 | [17] | ||
| Phintella cavaleriei | MW540530 | [20] | |||
| Dendryphantini | Dendryphantes sp. | MW832855 | [17] | ||
| Euophryini | Corythalia opima | OQ281589 | This study | ||
| Parabathippus shelfordi | OQ429315 | This study | |||
| Plexippini | Colopsus longipalpis | MH891570 | [26] | ||
| Epeus alboguttatus | MH922026 | [21] | |||
| Habronattus oregonensis | NC005942 | [22] | |||
| Plexippus paykulli | KM114572 | [27] | |||
| Telamonia vlijmi | NC024287 | [28] | |||
| Salticini | Carrhotus xanthogramma | KP402247 | [17,29] | ||
| Selenopidae | Selenops bursarius | NC024878 | [30] | ||
| Trochanteriidae | Plator insolens | OM397542 | [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Lin, L.; Ding, Y.; Zhang, F.; Zhang, J. Comparative Mitogenomics of Jumping Spiders with First Complete Mitochondrial Genomes of Euophryini (Araneae: Salticidae). Insects 2023, 14, 517. https://doi.org/10.3390/insects14060517
Zhang W, Lin L, Ding Y, Zhang F, Zhang J. Comparative Mitogenomics of Jumping Spiders with First Complete Mitochondrial Genomes of Euophryini (Araneae: Salticidae). Insects. 2023; 14(6):517. https://doi.org/10.3390/insects14060517
Chicago/Turabian StyleZhang, Wenqiang, Long Lin, Yuhui Ding, Feng Zhang, and Junxia Zhang. 2023. "Comparative Mitogenomics of Jumping Spiders with First Complete Mitochondrial Genomes of Euophryini (Araneae: Salticidae)" Insects 14, no. 6: 517. https://doi.org/10.3390/insects14060517