Development of Enzyme-Linked Immunosorbent and Immunochromatography Assays for Diagnosing Nosema ceranae Infection in Honey Bees


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of N. ceranae Spores (Preparation of N. ceranae Spore Antigens)
2.2. Monoclonal Antibody for N. ceranae
2.3. Feasibility Test of Antibody
2.4. Biotinylation of mAbs
2.5. SDS-PAGE
2.6. Sandwich ELISA
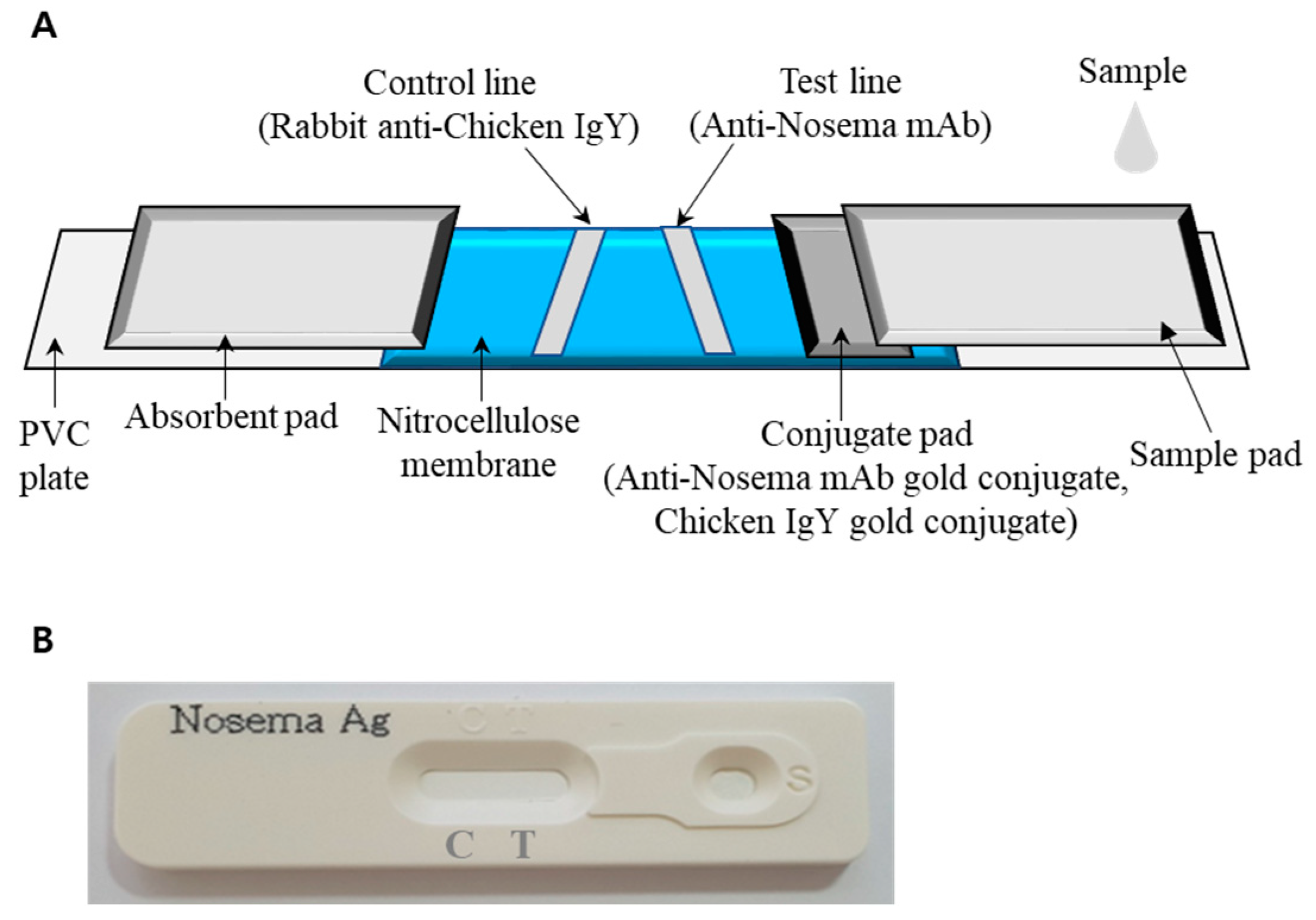
2.7. Immunochromatography (ICG) to Diagnose N. ceranae Infection
2.8. Pairing test of Colloidal Gold Strip
2.9. Detection of Nosema Spores in Experimental Specimens
2.10. Statistical Analysis
3. Results
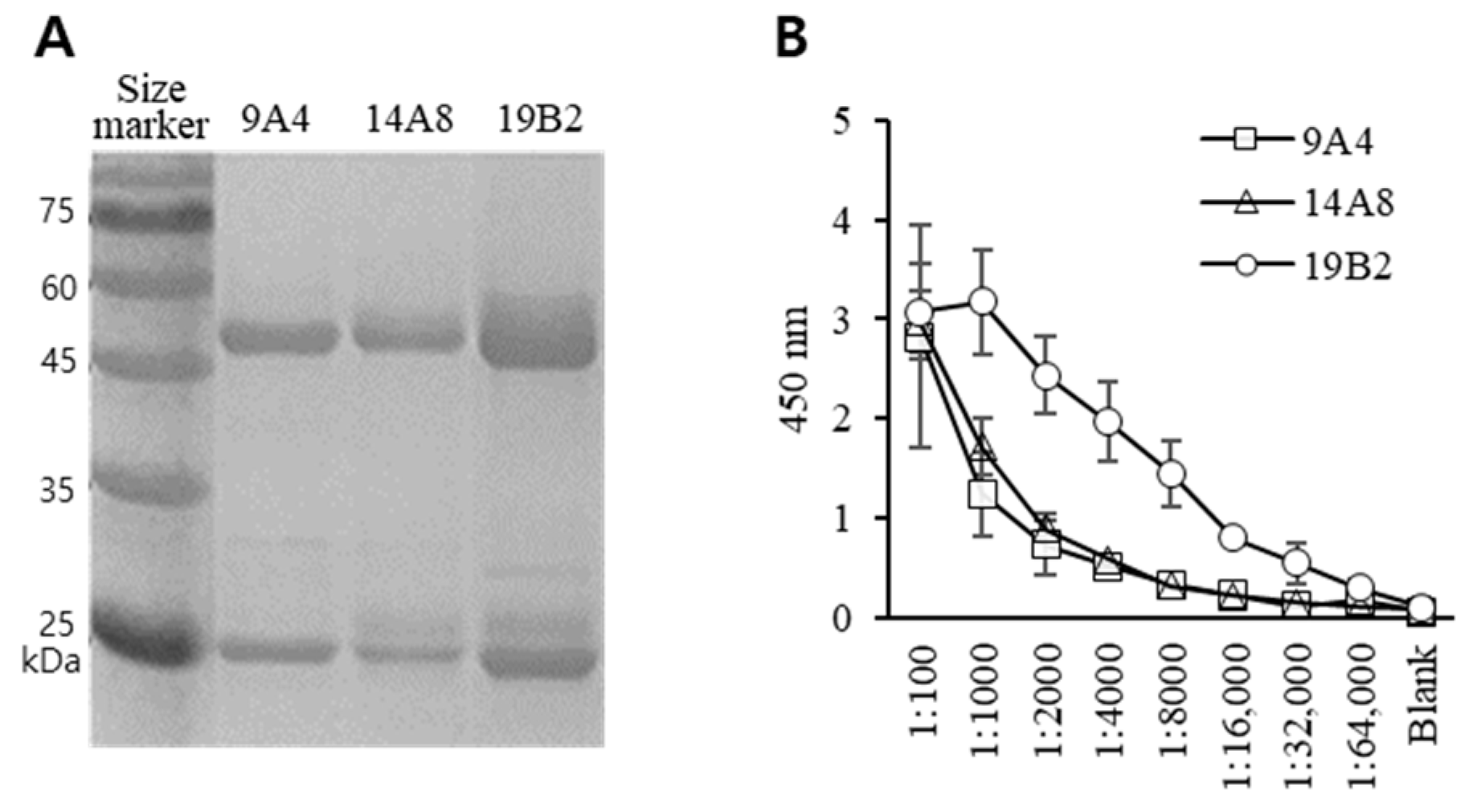
3.1. Generation of Hybridoma and Characterization of mAbs
3.2. Feasibility Test of Antibody
3.3. Effect of mAb Biotinylation on Antibody Binding to Nosema Spore
3.4. Sandwich ELISA for Diagnosis of Nosema Infection
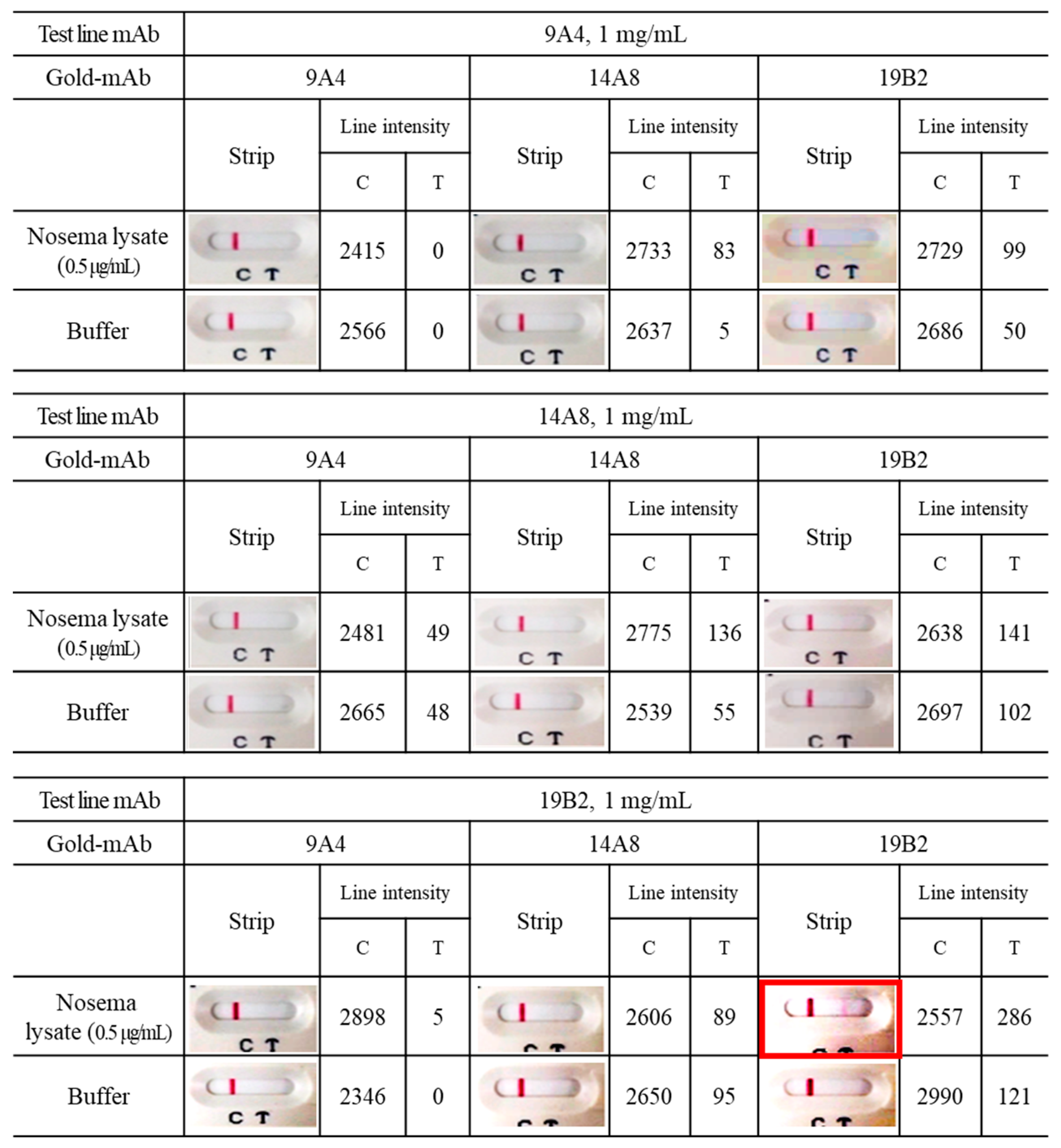
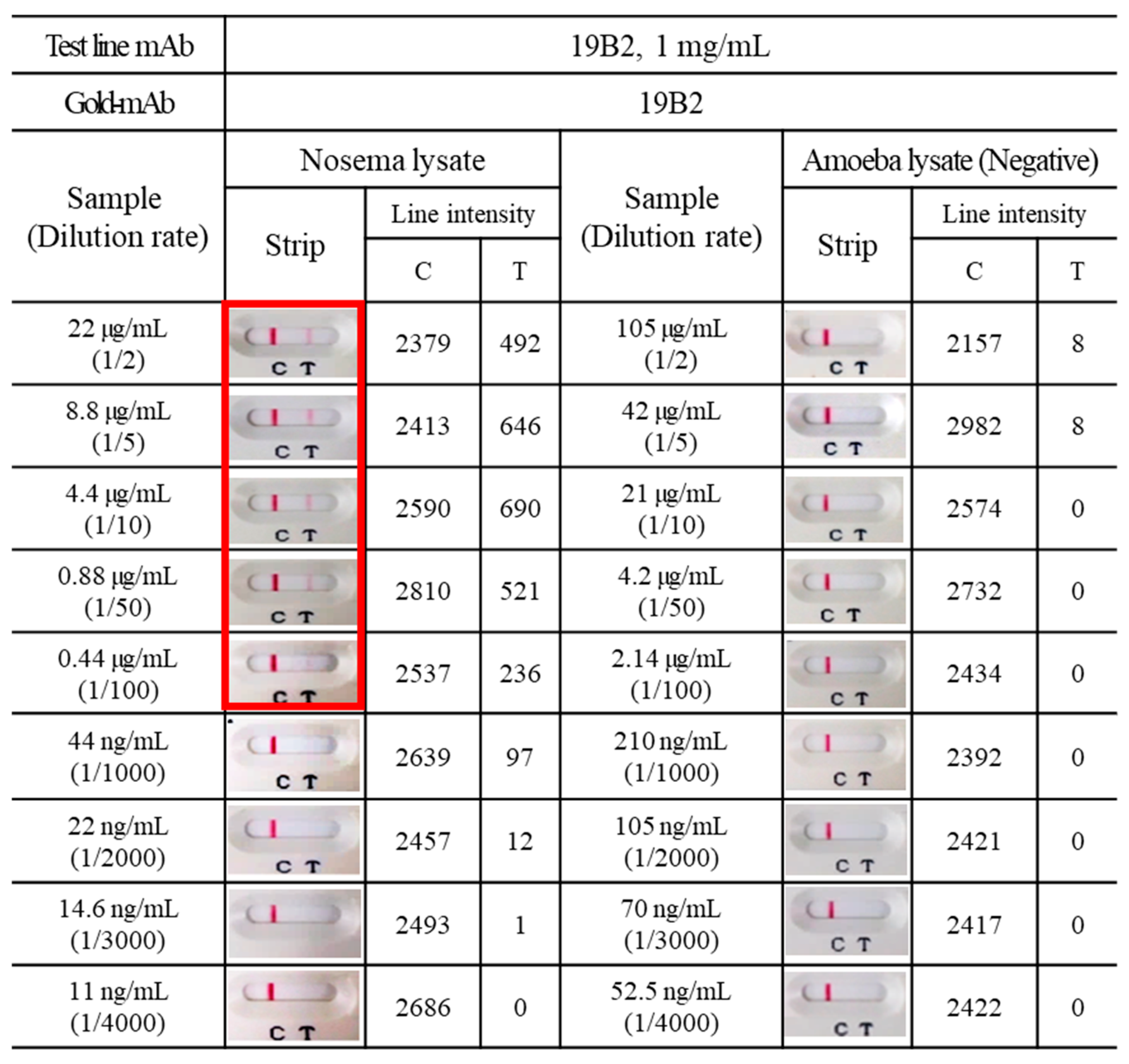
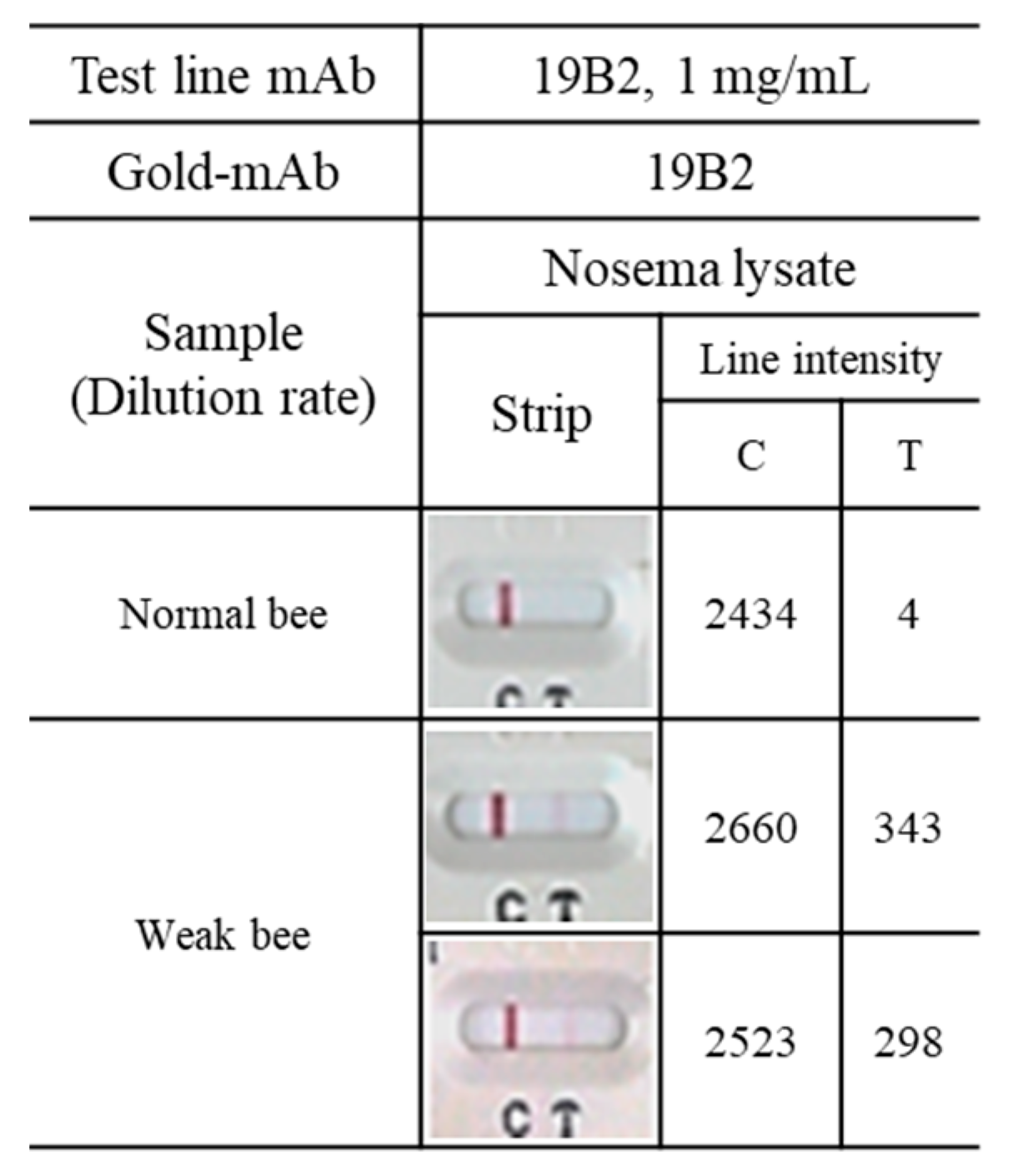
3.5. Application of mAbs to ICG
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fries, I.; Feng, F.; Dasilva, A.; Slemenda, S.B.; Pieniazek, N.J. Nosema ceranae n sp (Microspora, Nosematidae), morphological and molecular characterization of a microsporidian parasite of the Asian honey bee Apis cerana (Hymenoptera, Apidae). Eur. J. Protistol. 1996, 32, 356–365. [Google Scholar] [CrossRef]
- Higes, M.; Martin-Hernandez, R.; Botias, C.; Bailon, E.G.; Gonzalez-Porto, A.V.; Barrios, L.; Del Nozal, M.J.; Bernal, J.L.; Jimenez, J.J.; Palencia, P.G.; et al. How natural infection by Nosema ceranae causes honeybee colony collapse. Environ. Microbiol. 2008, 10, 2659–2669. [Google Scholar] [CrossRef]
- Klee, J.; Besana, A.M.; Genersch, E.; Gisder, S.; Nanetti, A.; Tam, D.Q.; Chinh, T.X.; Puerta, F.; Ruz, J.M.; Kryger, P.; et al. Widespread dispersal of the microsporidian Nosema ceranae, an emergent pathogen of the western honey bee, Apis mellifera. J. Invertebr. Pathol. 2007, 96, 1–10. [Google Scholar] [CrossRef]
- Paxton, R.J.; Klee, J.; Korpela, S.; Fries, I. Nosema ceranae has infected Apis mellifera in Europe since at least 1998 and may be more virulent than Nosema apis. Apidologie 2007, 38, 558–565. [Google Scholar] [CrossRef]
- Fries, I. Nosema ceranae in European honey bees (Apis mellifera). J. Invertebr. Pathol. 2010, 103, S73–S79. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, R.; Botías, C.; Bailón, E.G.; Martínez-Salvador, A.; Prieto, L.; Meana, A.; Higes, M. Microsporidia infecting Apis mellifera: Coexistence or competition. Is Nosema ceranae replacing Nosema apis? Environ. Microbiol. 2012, 14, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, M.; Sokot, R. Nosemosis in honey bees. Pol. J. Nat. Sci. 2014, 29, 91–99. [Google Scholar]
- Cantwell, G.E. Standard methods for counting nosema spores. Am. Bee J. 1970, 110, 222–223. [Google Scholar]
- Kim, J.H.; Park, J.K.; Lee, J.K. Evaluation of antimicrosporidian activity of plant extracts on Nosema ceranae. J. Apic. Sci. 2016, 60, 167–178. [Google Scholar] [CrossRef]
- Klee, J.; Tek Tay, W.; Paxton, R.J. Specific and sensitive detection of Nosema bombi (Microsporidia: Nosematidae) in bumble bees (Bombus spp.; Hymenoptera: Apidae) by PCR of partial rRNA gene sequences. J. Invertebr. Pathol. 2006, 91, 98–104. [Google Scholar] [CrossRef]
- Carletto, J.; Blanchard, P.; Gauthier, A.; Schurr, F.; Chauzat, M.P.; Ribiere, M. Improving molecular discrimination of Nosema apis and Nosema ceranae. J. Invertebr. Pathol. 2013, 113, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Lee, J.K. Development of monoclonal antibodies against spores of Nosema ceranae for the diagnosis of nosemosis. J. Apic. Res. 2021, 2022, 1–8. [Google Scholar] [CrossRef]
- Gisder, S.; Mockel, N.; Linde, A.; Genersch, E. A cell culture model for Nosema ceranae and Nosema apis allows new insights into the life cycle of these important honey bee-pathogenic microsporidia. Environ. Microbiol. 2011, 13, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Gisder, S.; Hedtke, K.; Mockel, N.; Frielitz, M.C.; Linde, A.; Genersch, E. Five-year cohort study of Nosema spp. in Germany: Does climate shape virulence and assertiveness of Nosema ceranae? Appl. Environ. Microbiol. 2010, 76, 3032–3038. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E.; Evans, J.D.; Fries, I. Honey bee disease overview. J. Invertebr. Pathol. 2010, 103 (Suppl. 1), S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Anuracpreeda, P.; Wanichanon, C.; Chawengkirtikul, R.; Chaithirayanon, K.; Sobhon, P. Fasciola gigantica: Immunodiagnosis of fasciolosis by detection of circulating 28.5 kDa tegumental antigen. Exp. Parasitol. 2009, 123, 334–340. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nautre 1970, 15, 680–685. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Qian, Y.; Guo, X.; Hu, X. Distinguishing the Pebrine spores N. bombycis, of the mulberry silkworm by immunoenzymatic technique. Sci. Seric. 1986, 72, 183–184. [Google Scholar]
- Greenstone, M.H. An Enzyme-Linked Immunosorbent-Assay for the Amblyospora Sp of Culex-Salinarius (Microspora, Amblyosporidae). J. Invertebr. Pathol. 1983, 41, 250–255. [Google Scholar] [CrossRef]
- Kawarabata, T.; Hayasaka, S. An Enzyme-Linked-Immunosorbent-Assay to Detect Alkali-Soluble Spore Surface-Antigens of Strains of Nosema-Bombycis (Microspora, Nosematidae). J. Invertebr. Pathol. 1978, 50, 118–123. [Google Scholar] [CrossRef]
- Aronstein, K.A.; Webster, T.C.; Saldivar, E. A serological method for detection of Nosema ceranae. J. Appl. Microbiol. 2013, 114, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-M.; Ree, H.J. Self-sandwich Method. An Improved Immunoperoxidase Technic for the Detection of Small Amounts of Antigens. Am. J. Clin. Pathol. 1980, 74, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Mertens, P.; De Vos, N.; Martiny, D.; Jassoy, C.; Mirazimi, A.; Cuypers, L.; Van den Wijngaert, S.; Monteil, V.; Melin, P.; Stoffels, K.; et al. Development and potential usefulness of the COVID-19 Ag Respi-Strip diagnostic assay in a pandemic context. Front. Med. 2020, 7, 225. [Google Scholar] [CrossRef]
- Korenkov, M.; Poopalasingam, N.; Madler, M.; Vanshylla, K.; Eggeling, R.; Wirtz, M.; Fish, I.; Dewald, F.; Gieselmann, L.; Lehmann, C.; et al. Evaluation of a rapid antigen test to detect SARS-CoV-2 infection and identify potentially infectious individuals. J. Clin. Microbiol. 2021, 59, e0089621. [Google Scholar] [CrossRef]
- Lippi, G.; Simundic, A.-M.; Plebani, M. Potential preanalytical and analytical vulnerabilities in the laboratory diagnosis of coronavirus disease 2019 (COVID-19). Clin. Chem. Lab. Med. 2020, 58, 1070–1076. [Google Scholar] [CrossRef]
- Richard-Greenblatt, M.; Ziegler, M.J.; Bromberg, V.; Huang, E.; Abdallah, H.; Tolomeo, P.; Lautenbach, E.; Glaser, L.; Kelly, B.J. Impact of nasopharyngeal specimen quality on SARS-CoV-2 test sensitivity. Med. Rxiv. 2020; preprint. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coated Antigen | Nosema Spore Lysate (1 μg/mL) | ||||||
|---|---|---|---|---|---|---|---|
| Primary Antibody | 9A4 | 14A8 | 19B2 | 9A4-biotin | 14A8-biotin | 19B2-biotin | |
| Antibody Concentration (ng/mL) | 1000 | 0.17 ± 0.015 1 | 1.99 ± 0.028 | 0.25 ± 0.01 | 0.27 ± 0.027 | 1.75 ± 0.296 | 1.07 ± 0.048 |
| 100 | 0.11 ± 0.005 | 0.33 ± 0.023 | 0.11 ± 0.026 | 0.24 ± 0.026 | 0.36 ± 0.047 | 0.34 ± 0.016 | |
| 10 | 0.11 ± 0.002 | 0.12 ± 0.002 | 0.09 ± 0.015 | 0.23 ± 0.015 | 0.26 ± 0.035 | 0.23 ± 0.012 | |
| 0 | 0.11 ± 0.006 | 0.09 ± 0.008 | 0.09 ± 0.012 | 0.22 ± 0.012 | 0.23 ± 0.015 | 0.25 ± 0.036 | |
| 2nd Antibody | Anti-mouse IgG Fc-HRP (1:10,000) | Avidin-HRP (1:5000) | |||||
| Coated Antigen | Nosema Spore (1 × 105 Cells/mL) | ||||||
|---|---|---|---|---|---|---|---|
| Primary Antibody | 9A4 | 14A8 | 19B2 | 9A4-biotin | 14A8-biotin | 19B2-biotin | |
| Antibody Concentration (ng/mL) | 1000 | 0.12 ± 0.011 1 | 0.37 ± 0.037 | 0.14 ± 0.007 | 0.22 ± 0.024 | 0.51 ± 0.021 | 0.42 ± 0.036 |
| 100 | 0.1 ± 0.012 | 0.11 ± 0.007 | 0.08 ± 0.003 | 0.19 ± 0.017 | 0.25 ± 0.033 | 0.25 ± 0.015 | |
| 10 | 0.08 ± 0.001 | 0.08 ± 0.004 | 0.07 ± 0.006 | 0.18 ± 0.011 | 0.22 ± 0.013 | 0.26 ± 0.03 | |
| 0 | 0.08 ± 0.003 | 0.09 ± 0.008 | 0.08 ± 0.004 | 0.21 ± 0.013 | 0.2 ± 0.003 | 0.19 ± 0.002 | |
| 2nd Antibody | Anti-mouse IgG Fc-HRP (1:10,000) | Avidin-HRP (1:5000) | |||||
| Capture Antibody | 9A4 Antibody (1 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore lysate (ng/mL) | 1000 | 0.20 ± 0.028 1 | 0.18 ± 0.006 | 0.22 ± 0.027 |
| 250 | 0.18 ± 0.017 | 0.16 ± 0.003 | 0.20 ± 0.022 | |
| Capture Antibody | 14A8 Antibody (1 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore lysate (ng/mL) | 1000 | 0.20 ± 0.016 | 0.22 ± 0.020 | 0.19 ± 0.006 |
| 250 | 0.18 ± 0.016 | 0.19 ± 0.008 | 0.18 ± 0.008 | |
| Capture Antibody | 19B2 Antibody (1 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore lysate (ng/mL) | 1000 | 0.18 ± 0.005 | 0.31 ± 0.096 | 0.21 ± 0.004 |
| 250 | 0.15 ± 0.011 | 0.15 ± 0.006 | 0.18 ± 0.014 | |
| Capture Antibody | 9A4 (5 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore Lysate (ng/mL) | 10,000 | 0.25 ± 0.029 1 | 0.39 ± 0.037 | 0.49 ± 0.011 |
| 2500 | 0.24 ± 0.010 | 0.31 ± 0.020 | 0.31 ± 0.015 | |
| 625 | 0.24 ± 0.018 | 0.29 ± 0.016 | 0.26 ± 0.011 | |
| 156.3 | 0.22 ± 0.012 | 0.29 ± 0.032 | 0.24 ± 0.013 | |
| 0 | 0.19 ± 0.005 | 0.26 ± 0.006 | 0.26 ± 0.015 | |
| Capture Antibody | 14A8 (5 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore Lysate (ng/mL) | 10,000 | 0.57 ± 0.020 | 2.88 ± 0.082 | 2.32 ± 0.093 |
| 2500 | 0.37 ± 0.037 | 1.03 ± 0.021 | 0.89 ± 0.112 | |
| 625 | 0.32 ± 0.010 | 0.56 ± 0.050 | 0.42 ± 0.011 | |
| 156.3 | 0.39 ± 0.033 | 0.42 ± 0.047 | 0.34 ± 0.029 | |
| 0 | 0.31 ± 0.017 | 0.34 ± 0.013 | 0.29 ± 0.015 | |
| Capture Antibody | 19B2 (5 μg/mL) | |||
| Detector Antibody | 9A4-biotin (1 μg/mL) | 14A8-biotin (1 μg/mL) | 19B2-biotin (1 μg/mL) | |
| Nosema spore Lysate (ng/mL) | 10,000 | 1.69 ± 0.090 | 3.04 ± 0.051 | 3.05 ± 0.055 |
| 2500 | 0.59 ± 0.020 | 2.53 ± 0.348 | 2.65 ± 0.117 | |
| 625 | 0.33 ± 0.011 | 0.97 ± 0.063 | 1.43 ± 0.088 | |
| 156.3 | 0.26 ± 0.015 | 0.39 ± 0.011 | 0.50 ± 0.038 | |
| 0 | 0.14 ± 0.052 | 0.23 ± 0.012 | 0.21 ± 0.007 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.K. Development of Enzyme-Linked Immunosorbent and Immunochromatography Assays for Diagnosing Nosema ceranae Infection in Honey Bees. Insects 2024, 15, 59. https://doi.org/10.3390/insects15010059
Lee JK. Development of Enzyme-Linked Immunosorbent and Immunochromatography Assays for Diagnosing Nosema ceranae Infection in Honey Bees. Insects. 2024; 15(1):59. https://doi.org/10.3390/insects15010059
Chicago/Turabian StyleLee, Jae Kwon. 2024. "Development of Enzyme-Linked Immunosorbent and Immunochromatography Assays for Diagnosing Nosema ceranae Infection in Honey Bees" Insects 15, no. 1: 59. https://doi.org/10.3390/insects15010059
APA StyleLee, J. K. (2024). Development of Enzyme-Linked Immunosorbent and Immunochromatography Assays for Diagnosing Nosema ceranae Infection in Honey Bees. Insects, 15(1), 59. https://doi.org/10.3390/insects15010059