Symbiotic Bacterial Communities of Insects Feeding on the Same Plant Lineage: Distinct Composition but Congruent Function



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Validation
2.3. 16S rRNA Gene Amplification and Sequencing
2.4. Sequencing Data Analysis
2.5. Diversity and Statistical Analysis
3. Results
3.1. Analysis of 16S rDNA Sequencing
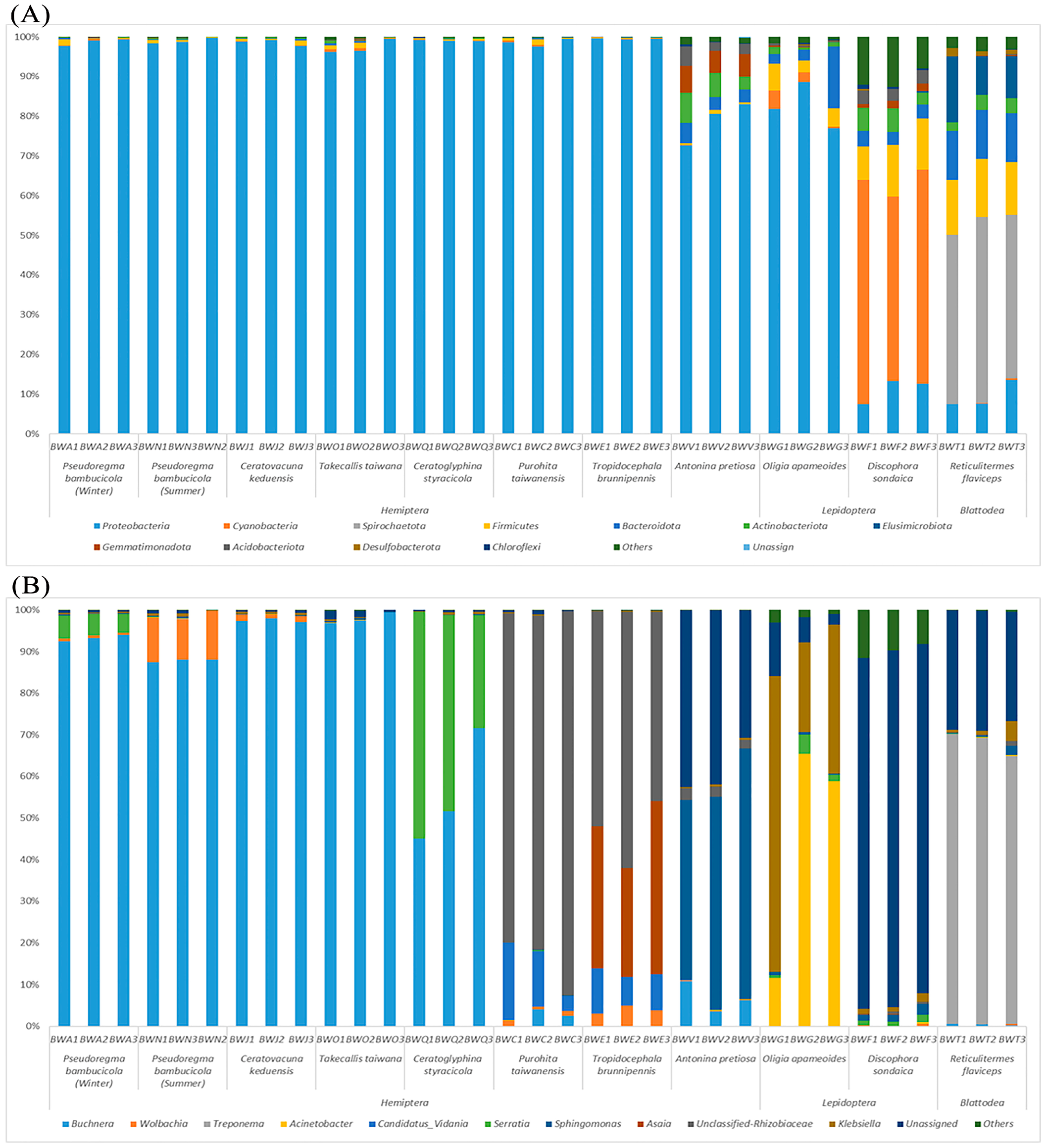
3.2. OTUs Distribution and Comparison of Symbionts between Diets and Phylogeny
3.3. Alpha Diversity
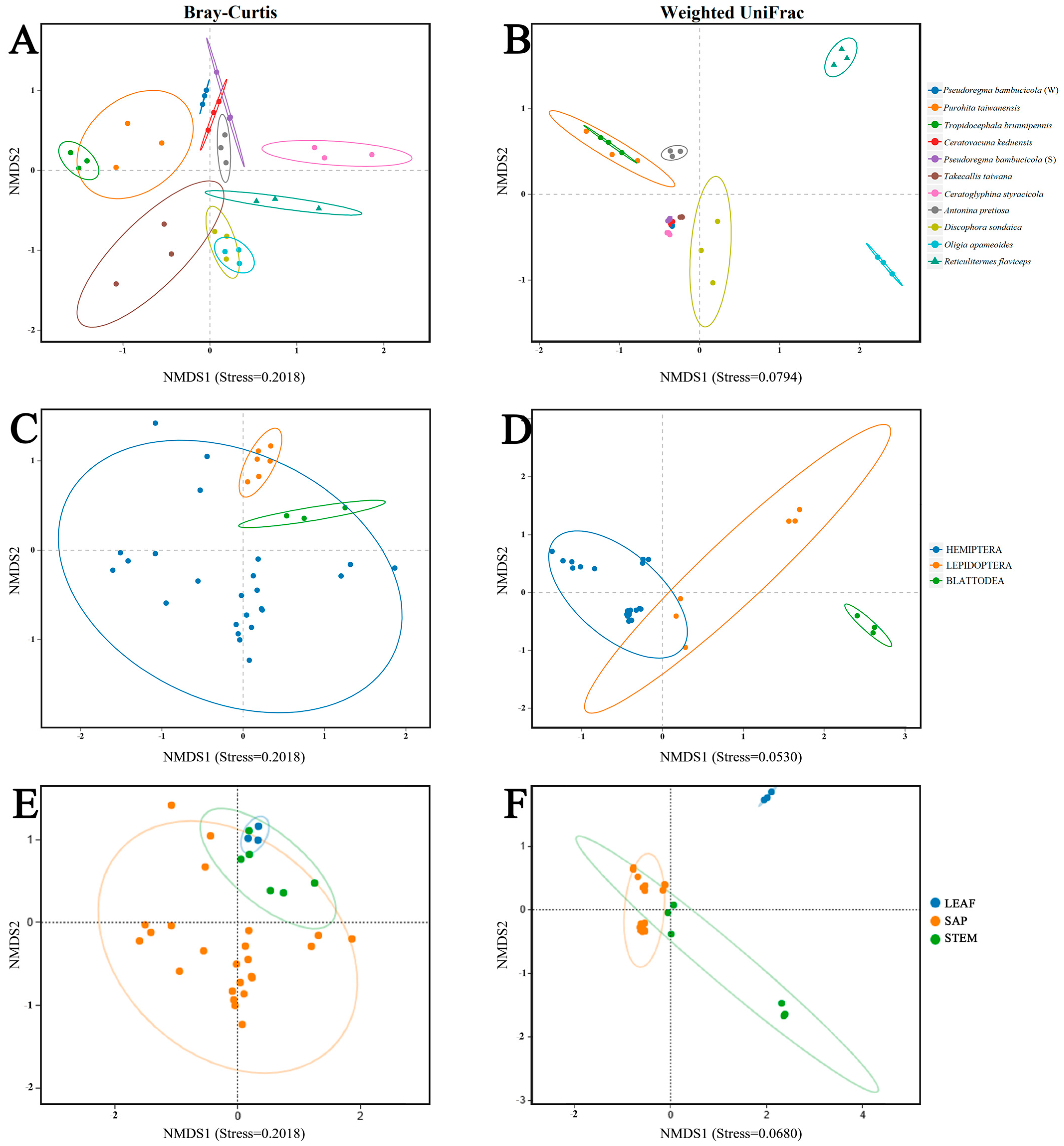
3.4. Beta Diversity
3.5. Functional Prediction of Bacterial Symbionts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yi, Z.; Jinchao, F.; Dayuan, X.U.E.; Weiguo, S.; Axmacher, J. Insect diversity: Addressing an important but strongly neglected research topic in China. J. Resour. Ecol. 2011, 2, 380–384. [Google Scholar]
- Douglas, A.E. The B vitamin nutrition of insects: The contributions of diet, microbiome and horizontally acquired genes. Curr. Opin. Insect Sci. 2017, 23, 65–69. [Google Scholar] [CrossRef]
- Afzal, W.; Junaid, N.; Siddiqui, A.; Longqin, Y.E.; Xiaolei, H. Functional interactions among phytophagous insects plants and microbial symbionts. WUYI Sci. J. 2020, 36, 1–15. [Google Scholar]
- Liu, Q.; Zhang, H.; Huang, X. Strong Linkage Between Symbiotic Bacterial Community and Host Age and Morph in a Hemipteran Social Insect. Microb. Ecol. 2022, 86, 1213–1225. [Google Scholar] [CrossRef]
- Hosokawa, T.; Koga, R.; Kikuchi, Y.; Meng, X.-Y.; Fukatsu, T. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. USA 2010, 107, 769–774. [Google Scholar] [CrossRef]
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef]
- Akman, L.; Yamashita, A.; Watanabe, H.; Oshima, K.; Shiba, T.; Aksoy, S. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 2002, 32, 402–407. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, K.; Li, Y.; Su, W.; Hu, K.; Jin, S. Enterococci mediate the oviposition preference of Drosophila melanogaster through sucrose catabolism. Sci. Rep. 2017, 7, 13420. [Google Scholar] [CrossRef]
- Hosokawa, T.; Kikuchi, Y.; Shimada, M.; Fukatsu, T. Symbiont acquisition alters behaviour of stinkbug nymphs. Biol. Lett. 2008, 4, 45–48. [Google Scholar] [CrossRef]
- Akami, M.; Andongma, A.A.; Zhengzhong, C.; Nan, J.; Khaeso, K.; Jurkevitch, E.; Niu, C.-Y.; Yuval, B. Intestinal bacteria modulate the foraging behavior of the oriental fruit fly Bactrocera dorsalis (Diptera: Tephritidae). PLoS ONE 2019, 14, e0210109. [Google Scholar] [CrossRef]
- Sepúlveda, D.A.; Zepeda-Paulo, F.; Ramírez, C.C.; Lavandero, B.; Figueroa, C.C. Diversity, frequency, and geographic distribution of facultative bacterial endosymbionts in introduced aphid pests. Insect Sci. 2017, 24, 511–521. [Google Scholar] [CrossRef]
- Henry, L.M.; Peccoud, J.; Simon, J.-C.; Hadfield, J.D.; Maiden, M.J.C.; Ferrari, J.; Godfray, H.C.J. Horizontally transmitted symbionts and host colonization of ecological niches. Curr. Biol. 2013, 23, 1713–1717. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, H.; Zeng, L.; Yu, Y.; Lin, X.; Huang, X. Coexistence of three dominant bacterial symbionts in a social aphid and implications for ecological adaptation. Insects 2021, 12, 416. [Google Scholar] [CrossRef]
- Chandler, J.A.; Lang, J.M.; Bhatnagar, S.; Eisen, J.A.; Kopp, A. Bacterial communities of diverse Drosophila species: Ecological context of a host–microbe model system. PLoS Genet. 2011, 7, e1002272. [Google Scholar] [CrossRef] [PubMed]
- Sandeu, M.M.; Maffo, C.G.T.; Dada, N.; Njiokou, F.; Hughes, G.L.; Wondji, C.S. Seasonal variation of microbiota composition in Anopheles gambiae and Anopheles coluzzii in two different eco-geographical localities in Cameroon. Med. Vet. Entomol. 2022, 36, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Rojas, L.M.; Scully, E.D.; Trick, H.N.; Zhu, K.Y.; Smith, C.M. Comparative analyses of transcriptional responses of Dectes texanus LeConte (Coleoptera: Cerambycidae) larvae fed on three different host plants and artificial diet. Sci. Rep. 2021, 11, 11448. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wang, J.; Huang, J.; Zhang, S.; Vogler, A.P.; Liu, Q.; Li, Y.; Yang, M.; Li, Y.; Zhou, X. Host Phylogeny and Diet Shape Gut Microbial Communities Within Bamboo-Feeding Insects. Front. Microbiol. 2021, 12, 633075. [Google Scholar] [CrossRef]
- Martin, M.M. The evolution of cellulose digestion in insects. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1991, 333, 281–288. [Google Scholar]
- Delsuc, F.; Metcalf, J.L.; Wegener Parfrey, L.; Song, S.J.; González, A.; Knight, R. Convergence of gut microbiomes in myrmecophagous mammals. Mol. Ecol. 2014, 23, 1301–1317. [Google Scholar] [CrossRef]
- Brune, A. Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 2014, 12, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Godon, J.-J.; Arcemisbéhère, L.; Escudié, R.; Harmand, J.; Miambi, E.; Steyer, J.-P. Overview of the oldest existing set of substrate-optimized anaerobic processes: Digestive tracts. BioEnergy Res. 2013, 6, 1063–1081. [Google Scholar] [CrossRef]
- Lazuka, A.; Auer, L.; O’Donohue, M.; Hernandez-Raquet, G. Anaerobic lignocellulolytic microbial consortium derived from termite gut: Enrichment, lignocellulose degradation and community dynamics. Biotechnol. Biofuels 2018, 11, 284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, M.; Wang, R.; Ma, Z.; Long, D.; Mao, H.; Wen, J.; Bernard, L.A.; Beauchemin, K.A.; Tan, Z. Urea plus nitrate pretreatment of rice and wheat straws enhances degradation and reduces methane production in in vitro ruminal culture. J. Sci. Food Agric. 2018, 98, 5205–5211. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, P.J. Nutritional Ecology of the Ruminant; Cornell University Press: Portland, OR, USA, 1982. [Google Scholar]
- Abe, T.; Higashi, M. Cellulose centered perspective on terrestrial community structure. Oikos 1991, 60, 127–133. [Google Scholar] [CrossRef]
- McKenney, E.A.; Maslanka, M.; Rodrigo, A.; Yoder, A.D. Bamboo specialists from two mammalian orders (Primates, Carnivora) share a high number of low-abundance gut microbes. Microb. Ecol. 2018, 76, 272–284. [Google Scholar] [CrossRef]
- Wang, H.; Varma, R.V.; Tiansen, X. Insects of Bamboos in Asia. An illustrated Manual; Brill: Beijing, China, 1998. [Google Scholar]
- Frame, L.A.; Costa, E.; Jackson, S.A. Current explorations of nutrition and the gut microbiome: A comprehensive evaluation of the review literature. Nutr. Rev. 2020, 78, 798–812. [Google Scholar] [CrossRef]
- Ledbetter, R.N.; Connon, S.A.; Neal, A.L.; Dohnalkova, A.; Magnuson, T.S. Biogenic mineral production by a novel arsenic-metabolizing thermophilic bacterium from the Alvord Basin, Oregon. Appl. Environ. Microbiol. 2007, 73, 5928–5936. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hoffman, N.G.; Morgan, M.T.; Matsen, F.A.; Fiedler, T.L.; Hall, R.W.; Ross, F.J.; McCoy, C.O.; Bumgarner, R.; Marrazzo, J.M. Bacterial communities in women with bacterial vaginosis: High resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS ONE 2012, 7, e37818. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; Wagner, H.; et al. Package ‘Vegan’: Community Ecology Package; R Foundation: Vienna, Austria, 2015. [Google Scholar]
- Wickham, H. Package ‘ggplot2’: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational multivariate analysis of variance (PERMANOVA). Wiley Statsref Stat. Ref. Online 2017, 1–15. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, S.; Luo, J.-Y.; Wang, C.-Y.; Lv, L.-M.; Cui, J.-J. Bacterial communities of the cotton aphid Aphis gossypii associated with Bt cotton in northern China. Sci. Rep. 2016, 6, 22958. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, D.; He, H.; Wei, C. Bacterial diversity of bacteriomes and organs of reproductive, digestive and excretory systems in two cicada species (Hemiptera: Cicadidae). PLoS ONE 2017, 12, e0175903. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, X.; Chen, Z.; Wang, Z.; Lu, Y.; Cheng, D. The divergence in bacterial components associated with Bactrocera dorsalis across developmental stages. Front. Microbiol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Eiri, D.; Moran, N.A.; Rangel, J. Modulation of the honey bee queen microbiota: Effects of early social contact. PLoS ONE 2018, 13, e0200527. [Google Scholar] [CrossRef] [PubMed]
- Mariño, Y.A.; Ospina, O.E.; Verle Rodrigues, J.C.; Bayman, P. High diversity and variability in the bacterial microbiota of the coffee berry borer (Coleoptera: Curculionidae), with emphasis on Wolbachia. J. Appl. Microbiol. 2018, 125, 528–543. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Koga, R.; Shibao, H.; Matsumoto, T.; Fukatsu, T. Diversity and geographic distribution of secondary endosymbiotic bacteria in natural populations of the pea aphid, Acyrthosiphon pisum. Mol. Ecol. 2002, 11, 2123–2135. [Google Scholar] [CrossRef]
- Gauthier, J.P.; Outreman, Y.; Mieuzet, L.; Simon, J.C. Bacterial communities associated with host-adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS ONE 2015, 10, e0120664. [Google Scholar] [CrossRef]
- Xu, S.; Jiang, L.; Qiao, G.; Chen, J. Diversity of bacterial symbionts associated with Myzus persicae (Sulzer)(Hemiptera: Aphididae: Aphidinae) revealed by 16S rRNA Illumina sequencing. Microb. Ecol. 2021, 81, 784–794. [Google Scholar] [CrossRef]
- Huang, G.; Wang, X.; Hu, Y.; Wu, Q.; Nie, Y.; Dong, J.; Ding, Y.; Yan, L.; Wei, F. Diet drives convergent evolution of gut microbiomes in bamboo-eating species. Sci. China Life Sci. 2021, 64, 88–95. [Google Scholar] [CrossRef]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, S.; Zheng, F.; Wang, N. Intestinal Bacterial Diversity and Functional Analysis of Three Lepidopteran Corn Ear Worm Larvae. Insects 2022, 13, 740. [Google Scholar] [CrossRef]
- Schmid, R.B.; Lehman, R.M.; Lundgren, J.G. Sex-specific interactions of microbial symbioses on cricket dietary selection. Environ. Entomol. 2014, 43, 896–902. [Google Scholar] [CrossRef]
- Andongma, A.A.; Wan, L.; Dong, Y.C.; Li, P.; Desneux, N.; White, J.A.; Niu, C.Y. Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis. Sci. Rep. 2015, 5, 9470. [Google Scholar] [CrossRef]
- Gao, X.; Li, W.; Luo, J.; Zhang, L.; Ji, J.; Zhu, X.; Wang, L.; Zhang, S.; Cui, J. Biodiversity of the microbiota in Spodoptera exigua (Lepidoptera: Noctuidae). J. Appl. Microbiol. 2019, 126, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Ishak, H.D.; Plowes, R.; Sen, R.; Kellner, K.; Meyer, E.; Estrada, D.A.; Dowd, S.E.; Mueller, U.G. Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb. Ecol. 2011, 61, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yao, Z.; Zheng, W.; Zhang, H. Bacterial communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 2014, 9, e106988. [Google Scholar] [CrossRef]
- Arora, J.; Kinjo, Y.; Šobotník, J.; Buček, A.; Clitheroe, C.; Stiblik, P.; Roisin, Y.; Žifčáková, L.; Park, Y.C.; Kim, K.Y.; et al. The functional evolution of termite gut microbiota. Microbiome 2022, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Sheehan, T.H.; Mott, B.M.; Maes, P.; Snyder, L.; Schwan, M.R.; Walton, A.; Jones, B.M.; Corby-Harris, V. Microbial ecology of the hive and pollination landscape: Bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PLoS ONE 2013, 8, e83125. [Google Scholar] [CrossRef] [PubMed]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2019, 13, 576–587. [Google Scholar] [CrossRef]
- Aylward, F.O.; Suen, G.; Biedermann, P.H.W.; Adams, A.S.; Scott, J.J.; Malfatti, S.A.; Glavina del Rio, T.; Tringe, S.G.; Poulsen, M.; Raffa, K.F. Convergent bacterial microbiotas in the fungal agricultural systems of insects. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef]
- Mikaelyan, A.; Dietrich, C.; Köhler, T.; Poulsen, M.; Sillam-Dussès, D.; Brune, A. Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol. Ecol. 2015, 24, 5284–5295. [Google Scholar] [CrossRef]
- Wang, Q.; Yuan, E.; Ling, X.; Zhu-Salzman, K. An aphid facultative symbiont suppresses plant defence by manipulating aphid gene expression in salivary glands. Plant Cell Environ. 2020, 43, 2311–2322. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, H.; Zhao, H.; Xian, Y.; Guo, H.; Liu, B.; Xue, K. Microbiome diversity of cotton aphids (Aphis gossypii) is associated with host alternation. Sci. Rep. 2021, 11, 5260. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, J.; Chen, M.; Yang, Q.; Du, X.; Chen, S.; Zhang, L.; Yu, Y.; Yu, W. Wolbachia infection status and genetic structure in natural populations of Polytremis nascens (Lepidoptera: Hesperiidae). Infect. Genet. Evol. 2014, 27, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.P.; Windsor, D.M.; Saucedo, J.M.; Werren, J.H. Reproductive effects and geographical distributions of two Wolbachia strains infecting the Neotropical beetle, Chelymorpha alternans Boh.(Chrysomelidae, Cassidinae). Mol. Ecol. 2004, 13, 2405–2420. [Google Scholar] [CrossRef]
- Martins, C.; Souza, R.F.; Bueno, O.C. Presence and distribution of the endosymbiont Wolbachia among Solenopsis spp.(Hymenoptera: Formicidae) from Brazil and its evolutionary history. J. Invertebr. Pathol. 2012, 109, 287–296. [Google Scholar] [CrossRef]
- Wei, T.; Ishida, R.; Miyanaga, K.; Tanji, Y. Seasonal variations in bacterial communities and antibiotic-resistant strains associated with green bottle flies (Diptera: Calliphoridae). Appl. Microbiol. Biotechnol. 2014, 98, 4197–4208. [Google Scholar] [CrossRef]
- Chen, R.; Su, X.; Chen, J.; Jiang, L.; Qiao, G.-X. Wolbachia infection in two species: Novel views on the colonization ability of Wolbachia in aphids. Environ. Entomol. 2019, 48, 1388–1393. [Google Scholar] [CrossRef]
- Chamankar, B.; Maleki-Ravasan, N.; Karami, M.; Forouzan, E.; Karimian, F.; Naeimi, S.; Choobdar, N. The structure and diversity of microbial communities in Paederus fuscipes (Coleoptera: Staphylinidae): From ecological paradigm to pathobiome. Microbiome 2023, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Gurung, K.; Wertheim, B.; Falcao Salles, J. The microbiome of pest insects: It is not just bacteria. Entomol. Exp. Appl. 2019, 167, 156–170. [Google Scholar] [CrossRef]
- Hansen, A.K.; Moran, N.A. The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol. Ecol. 2014, 23, 1473–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, R.; Liu, Q.; Lu, J.; Qiao, G.; Huang, X. Transcriptomic and proteomic analyses provide insights into host adaptation of a bamboo-feeding aphid. Front. Plant Sci. 2023, 13, 1098751. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No | Collection Date | Sample ID | Host Plant | Insect Specie | Insect Order | Feeding | Sample Name in Figures |
|---|---|---|---|---|---|---|---|
| 1 | 1 November 2022 | BWA1 | Bambusa sp. | Pseudoregma bambucicola (Winter) | Hemiptera | Sap | |
| 2 | BWA2 | PbW.Sa.H | |||||
| 3 | BWA3 | ||||||
| 4 | 4 May 2023 | BWC1 | Bambusa sp. | Purohita taiwanensis | Sap | ||
| 5 | BWC2 | Pt.Sa.H | |||||
| 6 | BWC3 | ||||||
| 7 | 6 May 2023 | BWE1 | Bambusa sp. | Tropidocephala brunnipennis | Sap | ||
| 8 | BWE2 | Tb.Sa.H | |||||
| 9 | BWE3 | ||||||
| 10 | 8 May 2023 | BWO1 | Bambusa sp. | Takecallis taiwana | Sap | ||
| 11 | BWO2 | Tt.Sa.H | |||||
| 12 | BWO3 | ||||||
| 13 | 9 May 2023 | BWJ1 | Bambusa sp. | Ceratovacuna keduensis | Sap | ||
| 14 | BWJ2 | Ck.Sa.H | |||||
| 15 | BWJ3 | ||||||
| 16 | 9 May 2023 | BWN1 | Bambusa sp. | Pseudoregma bambucicola (Summer) | Sap | ||
| 17 | BWN2 | PbS.Sa.H | |||||
| 18 | BWN3 | ||||||
| 19 | 9 May 2023 | BWQ1 | Bambusa sp. | Ceratoglyphina styracicola | Sap | ||
| 20 | BWQ2 | Ct.Sa.H | |||||
| 21 | BWQ3 | ||||||
| 22 | 4 June 2023 | BWV1 | Bambusa sp. | Antonina pretiosa | Sap | ||
| 23 | BWV2 | Ap.Sa.H | |||||
| 24 | BWV3 | ||||||
| 25 | 11 June 2023 | BWF1 | Bambusa sp. | Discophora sondaica | Lepidoptera | Leaf | |
| 26 | BWF2 | Ds.Lf.L | |||||
| 27 | BWF3 | ||||||
| 28 | 2 May 2023 | BWG1 | Bambusa sp. | Oligia apameoides | Stem | ||
| 29 | BWG2 | Oa.St.L | |||||
| 30 | BWG3 | ||||||
| 31 | 8 May 2023 | BWT1 | Bambusa sp. | Reticulitermes flaviceps | Blattodea | Stem | |
| 32 | BWT2 | Rf.St.B | |||||
| 33 | BWT3 |
| Beta Diversity Distance | Bacterial Community | ANOSIM (R, p) | PERMANOVA (R2, p) |
|---|---|---|---|
| Bray–Curtis | All Samples | 0.935, 0.001 | 0.936, 0.001 |
| Insect Orders | 0.223, 0.003 | 0.235, 0.001 | |
| Type of food (Sap, Leaf, Stem) | 0.189, 0.005 | 0.217, 0.001 | |
| Season (Summer, Winter) | 0.481, 0.01 | 0.240, 0.01 | |
| Weighted Unifrac | All Samples | 0.888, 0.001 | 0.970, 0.001 |
| Insect Orders | 0.721, 0.001 | 0.509, 0.001 | |
| Type of food (Sap, Leaf, Stem) | 0.718, 0.001 | 0.483, 0.001 | |
| Season (Summer, Winter) | 0.333, 0.02 | 0.174, 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naveed, W.A.; Liu, Q.; Lu, C.; Huang, X. Symbiotic Bacterial Communities of Insects Feeding on the Same Plant Lineage: Distinct Composition but Congruent Function. Insects 2024, 15, 187. https://doi.org/10.3390/insects15030187
Naveed WA, Liu Q, Lu C, Huang X. Symbiotic Bacterial Communities of Insects Feeding on the Same Plant Lineage: Distinct Composition but Congruent Function. Insects. 2024; 15(3):187. https://doi.org/10.3390/insects15030187
Chicago/Turabian StyleNaveed, Waleed Afzal, Qian Liu, Congcong Lu, and Xiaolei Huang. 2024. "Symbiotic Bacterial Communities of Insects Feeding on the Same Plant Lineage: Distinct Composition but Congruent Function" Insects 15, no. 3: 187. https://doi.org/10.3390/insects15030187