

Comparative Mitogenomics and Phylogenetic Implications for Nine Species of the Subfamily Meconematinae (Orthoptera: Tettigoniidae)
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Extraction and Sequencing
2.2. Mitochondrial Genome Sequence Screening and Assembly
2.3. Annotation and Analysis of the Mitochondrial Genome
2.4. Construction of Phylogenetic Trees
3. Results and Discussion
3.1. Comparison of Mitochondrial Genome Lengths
3.2. Mitochondrial Gene Interval and Overlapping Regions
3.3. Nucleotide Composition and Skew
3.4. Protein-Coding Genes and Codon Usage
3.5. tRNA and rRNA Genes
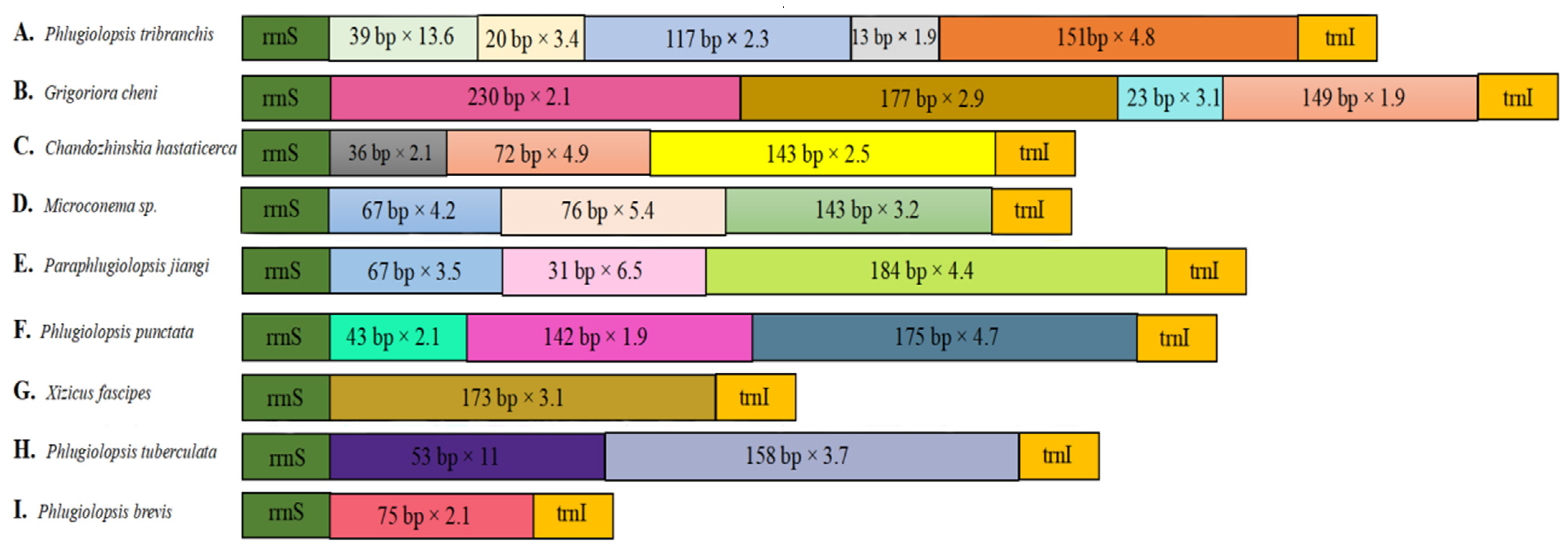
3.6. Control Region
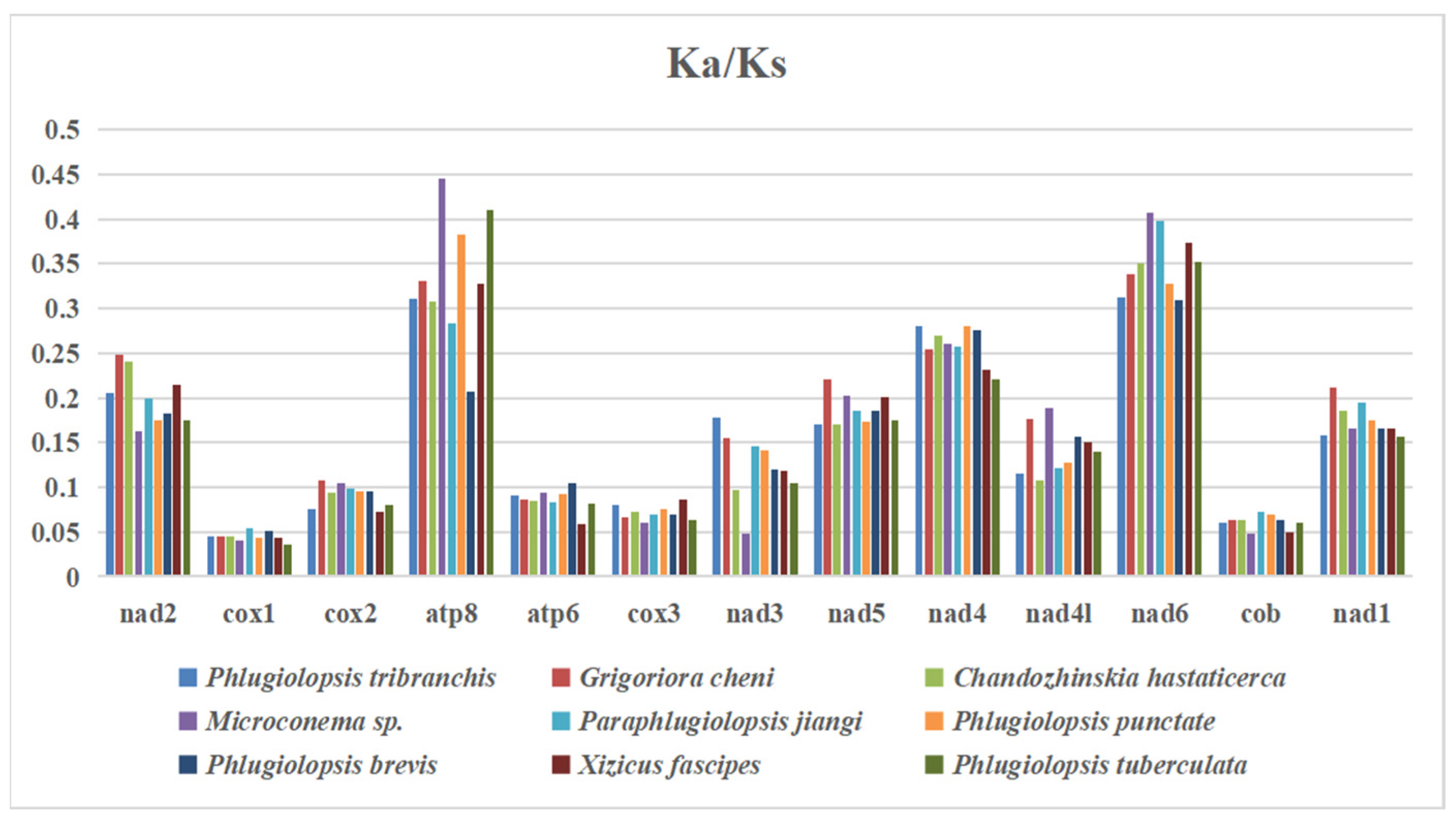
3.7. The Rate of Evolution of 13 PCGs
3.8. Substitution Saturation Tests and Nucleotide Heterogeneity
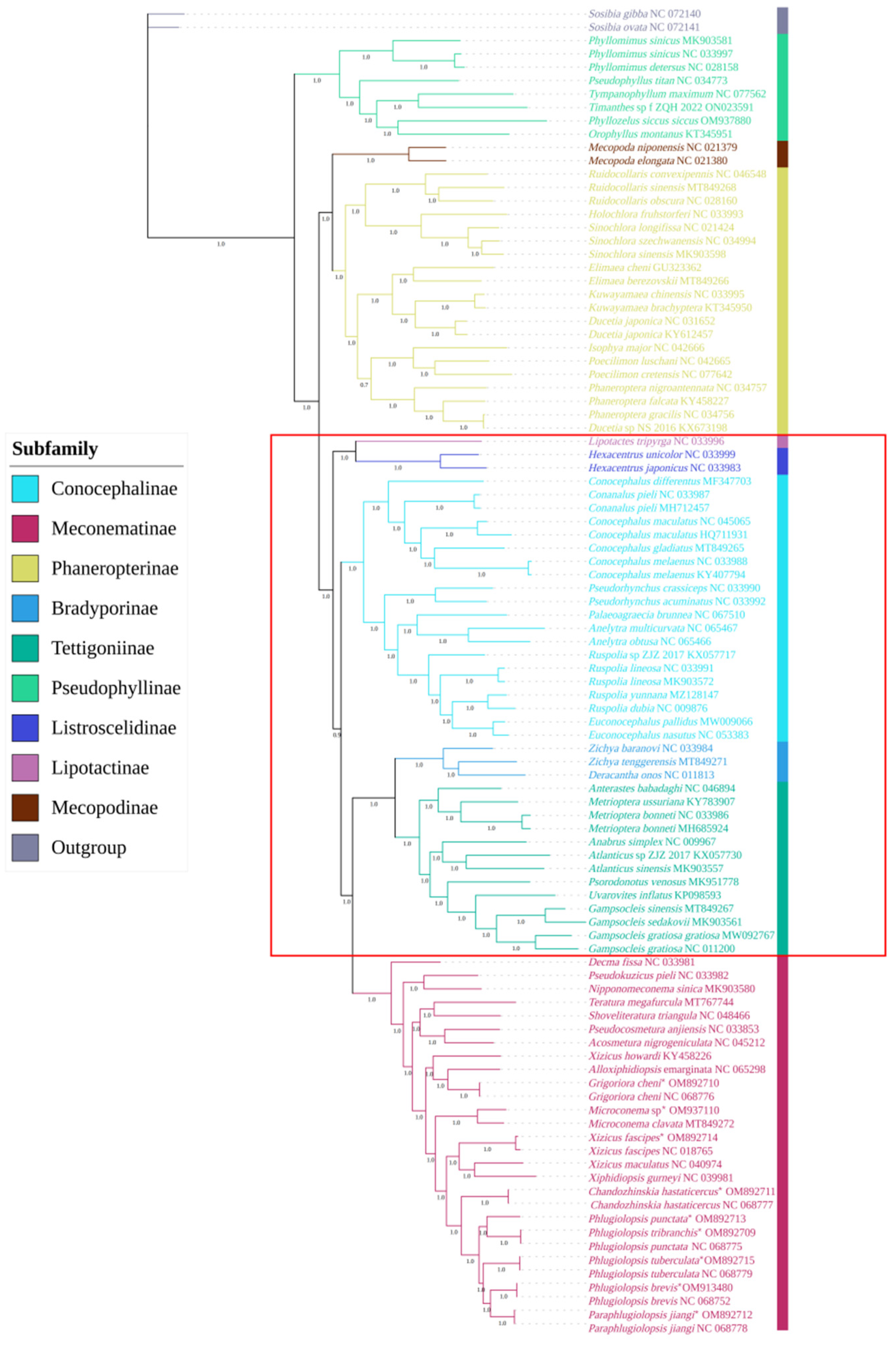
3.9. Phylogenetic Analysis of Meconematinae
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorochov, A.V. New and little known katydids of the tribe Meconematini (Orthoptera: Tettigoniidae: Meconematinae) from south-east Asia. Труды Зooлoгическoгo Института РАН 2008, 312, 26–42. [Google Scholar] [CrossRef]
- Burmeister, H. Kaukerfe, Gymnognatha (Erste Hälfte: Vulgo Orthoptera). Handb. Der Entomol. 1838, 2, 397–756. [Google Scholar]
- Mao, S.; Yuan, H.; Chang, H.; Shi, F.; Zhou, Y. Comparative mitochondrial genomics of Shoveliteratura triangula (Orthoptera, Tettigoniidae, Meconematinae) and the first description of a female specimen. Zootaxa 2020, 4751, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Gorochov, A.V.; Liu, C.; Kang, L. Studies on the tribe Meconematini (Orthoptera: Tettigoniidae: Meconematinae) from China. Orient Insects 2005, 39, 63–87. [Google Scholar] [CrossRef]
- Wang, H.; Jing, J.; Liu, X.; Li, K. Revision on genus Xizicus Gorochov (Orthoptera, Tettigoniidae, Meconematinae, Meconematini) with description of three new species form China. Zootaxa 2014, 3861, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Shi, F.; Chang, Y. Two new species of the genus Phlugiolopsis (Tettigoniidae: Meconematinae) and the male description of Phlugiolopsis circolobosis Bian, Shi & Chang, 2013 from Yunnan, China. Zootaxa 2023, 5296, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, H.; Shi, F. One new species of the genus Acosmetura (Orthoptera: Tettigoniidae: Meconematinae) from Hunan, China. Zootaxa 2023, 5369, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bian, X. One new species of Xizicus in the genus subgenus Paraxizicus (Orthoptera: Tettigoniidae: Meconematinae) from Yunnan Province, China. Zootaxa 2023, 5325, 147–150. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Chen, C.; Shi, F. One new genus and species of the tribe Meconematini (Orthoptera: Tettigoniidae: Meconematinae) from Guangxi, China. Zootaxa 2023, 5227, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Wang, T.; Shi, F. New genera and species of brachypterous members of the tribe Meconematini (Orthoptera: Tettigoniidae) from Hunan, China. Zootaxa 2020, 4819, 4819. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.P.; Paulay, G. DNA barcoding: Error rates based on comprehensive sampling. PLoS Biol. 2005, 3, e422. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Shiyang, K.; Vaidya, G.; Ng, P.K.L. DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 2006, 55, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Trewick, S.A. DNA Barcoding is not enough: Mismatch of taxonomy and genealogy in New Zealand grasshoppers (Orthoptera: Acrididae). Cladistics 2008, 24, 240–254. [Google Scholar] [CrossRef]
- Funk, D.J.; Omland, K.E. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 397–423. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhao, L.; Liu, N.; Guo, H.; Guan, B.; Di, J.; Shi, F. Towards a higher-level Ensifera phylogeny inferred from mitogenome sequences. Mol. Phylogenet. Evol. 2017, 108, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Macino, G.; Scazzocchio, C.; Waring, R.B.; Berks, M.M.P.; Wayne Davies, R. Conservation and rearrangement of mitochondrial structural gene sequences. Nature 1980, 288, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chang, J.; Ma, C.; Li, L.; Zhou, S. Mitochondrial genomes of two Sinochlora species (Orthoptera): Novel genome rearrangements and recognition sequence of replication origin. BMC Genom. 2013, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.L.; Li, X.J.; Zhang, H.L.; Zheng, Z.M. Mitochondrial genomes of three Tetrigoidea species and phylogeny of Tetrigoidea. PeerJ 2017, 5, e4002. [Google Scholar] [CrossRef] [PubMed]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.M.; George, J.M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Song, H.J.; Matthew, M.J.; Michael, M.F. Rampant nuclear insertion of mtDNA across diverse lineages within Orthoptera (Insecta). PLoS ONE 2014, 9, e110508. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Amédégnato, C.; Cigliano, M.M.; Desutter-Grandcolas, L.; Heads, S.W.; Huang, Y.; Otte, D.; Whiting, M.F. 300 million years of diversification: Elucidating the patterns of orthopteran evolution based on comprehensive taxon and gene sampling. Cladistics 2015, 31, 621–651. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.A.; Chiang, B.H. The Nearctic Nedubini: The most basal lineage of katydids is resolved among the paraphyletic “Tettigoniinae” (Orthoptera: Tettigoniidae). Ann. Entomol. Soc. Am. 2016, 109, 652–662. [Google Scholar] [CrossRef]
- Mao, S.L.; Lu, Y.; Xun, L.L.; Zhou, Y.F. Characterization of the mitochondrial genome of Alloxiphidiopsis emarginata (Orthoptera, Tettigoniidae, Meconematinae). Mitochondrial DNA Part B 2019, 4, 4192–4193. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.L.; Qiu, Z.Y.; Li, Q.; Li, Y.; Zhou, Y.F. Complete mitochondrial genome of Xiphidiopsis (Xiphidiopsis) gurneyi (Orthoptera, Tettigoniidae, Meconematinae). Mitochondrial DNA Part B 2018, 3, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.L.; Yuan, H.; Lu, C.; Zhou, Y.F.; Shi, F.M. The complete mitochondrial genome of Xizicus (Haploxizicus) maculatus revealed by next-generation sequencing and phylogenetic implication (Orthoptera, Meconematinae). ZooKeys 2018, 773, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.R.; Zhou, Z.J.; Chang, Y.L.; Zhao, L.H. The mitochondrial genome of the quiet-calling katydids, Xizicus fascipes (Orthoptera: Tettigoniidae: Meconematinae). J. Genet. 2012, 91, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, F. Mitogenome sequence analysis of Xizicus howardi (Orthoptera: Tettigoniidae). Genom. Appl. Biol. 2017, 36, 3194–3199. [Google Scholar]
- Han, N.; Yuan, H.; Wang, J.; Zhou, Y.; Mao, S. Mitochondrial genome of a brachypterous species in Meconematinae: Acosmetura nigrogeniculata and its phylogenetic implication. Mitochondrial DNA Part B 2019, 4, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Matvienko, M. CLC Genomics Workbench. Plant Anim. Genome Sr. Field Appl. Sci. CLC Bio 2015, 1, 1–42. [Google Scholar]
- NCBI. National Center for Biotechnology Information [EB/OL]. 2024. Available online: https://www.ncbi.nlm.nih.gov (accessed on 14 January 2024).
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE–visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Biol. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.H. DAMBE5: A comprehensive software package fordata analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van, D.M.P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von, H.A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Catanzaro, D.; Milinkovitch, M.C. Assessing the applicability of the GTR nucleotide substitution model through simulations. Evol. Bioinform. 2006, 2, 145–155. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Foerstner, K.U.; Mering, C.V.; Hooper, S.D.; Bork, P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005, 6, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Debrauwere, H.; Gendrel, C.G.; Lechat, S.; Dutreix, M. Differences and similarities between various tandem repeat sequences: Minisatellites and microsatellites. Biochimie 1997, 79, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C.; Brown, W.M. Tandem duplications in animal mitochondrial DNAs: Variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. USA 1987, 84, 7183–7187. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M. Concerted evolution and RAPping in mitochondrial VNTRs and the molecular geography of cricket populations. In Molecular Ecology and Evolution: Approaches and Applications; Birkhäuser: Basel, Switzerland, 1994; pp. 227–245. [Google Scholar] [CrossRef]
- Cai, J.J.; Borenstein, E.; Chen, R.; Petrov, D.A. Similarly strong purifying selection acts on human disease genes of all evolutionary ages. Genome Biol. Evol. 2009, 1, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.Y.; Nikolaev, S.I.; Junier, T.; Baranova, M.; Antonarakis, S.E. Purifying selection in mammalian mitochondrial protein-coding genes is highly effective and congruent with evolution of nuclear genes. Mol. Biol. Evol. 2012, 30, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Shi, P.; Sun, Y.B.; Zhang, Y.P. Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Res. 2009, 19, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Qiu, Z.; Yuan, H.; Wang, X.; Li, X.; Sun, H. Evolutionary rates of and selective constraints on the mitochondrial genomes of Orthoptera insects with different wing types. Mol. Phylogenet. Evol. 2020, 145, 106734. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.R.; Dowton, M. Mitochondrial genomes in the Hymenoptera and their utility as phylogenetic markers. Syst. Entomol. 2007, 32, 60–69. [Google Scholar] [CrossRef]
- Pons, J.; Ribera, I.; Bertranpetit, J.; Balke, M. Nucleotide substitution rates for the full set of mitochondrial protein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 2010, 56, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.E.; Breeschoten, T.; Timmermans, M.J.; Nadein, K.; Xue, H.J.; Bai, M.; Huang, Y.; Yang, X.K.; Vogler, A.P. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics 2018, 34, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Xu, W.; Zhang, D.; Li, J. Comparative analysis of the mitochondrial genomes of flesh flies and their evolutionary implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Y.; Wang, Y.L.; Liang, L.; Yin, Z.W.; Thayer, M.K.; Newton, A.F.; Zhou, Y.L. Congruence of morphological and molecular phylogenies of the rove beetle subfamily Staphylininae (Coleoptera: Staphylinidae). Sci. Rep. 2019, 9, 15137. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Xie, G.L.; Chang, Y.L.; Shi, F.M. One new genus and two new species of the tribe Meconematini (Orthoptera: Tettigoniidae: Meconematinae) from Yunnan, China. Zootaxa 2014, 3793, 286–290. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, S.; Zhang, Q.; Liang, L.; Qin, Y.; Li, S.; Bian, X. Comparative Mitogenomics and Phylogenetic Implications for Nine Species of the Subfamily Meconematinae (Orthoptera: Tettigoniidae). Insects 2024, 15, 413. https://doi.org/10.3390/insects15060413
Pang S, Zhang Q, Liang L, Qin Y, Li S, Bian X. Comparative Mitogenomics and Phylogenetic Implications for Nine Species of the Subfamily Meconematinae (Orthoptera: Tettigoniidae). Insects. 2024; 15(6):413. https://doi.org/10.3390/insects15060413
Chicago/Turabian StylePang, Siyu, Qianwen Zhang, Lili Liang, Yanting Qin, Shan Li, and Xun Bian. 2024. "Comparative Mitogenomics and Phylogenetic Implications for Nine Species of the Subfamily Meconematinae (Orthoptera: Tettigoniidae)" Insects 15, no. 6: 413. https://doi.org/10.3390/insects15060413
APA StylePang, S., Zhang, Q., Liang, L., Qin, Y., Li, S., & Bian, X. (2024). Comparative Mitogenomics and Phylogenetic Implications for Nine Species of the Subfamily Meconematinae (Orthoptera: Tettigoniidae). Insects, 15(6), 413. https://doi.org/10.3390/insects15060413