
Haplotype Diversity in mtDNA of Honeybee in the Czech Republic Confirms Complete Replacement of Autochthonous Population with the C Lineage


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling of Bees
2.2. DNA Extraction
2.3. PCR Amplification
2.4. DNA Sequencing
2.5. Data Analysis
3. Results
3.1. Analysis of tRNAleu-cox2 Sequences
3.2. Analysis of Polymorphisms of tRNAleu-cox2
3.3. Description of New Haplotypes
3.4. Analysis of cox1 Sequences
3.5. Distribution of Haplotypes Based on the Sampling Technique
3.6. Haplotype Diversity
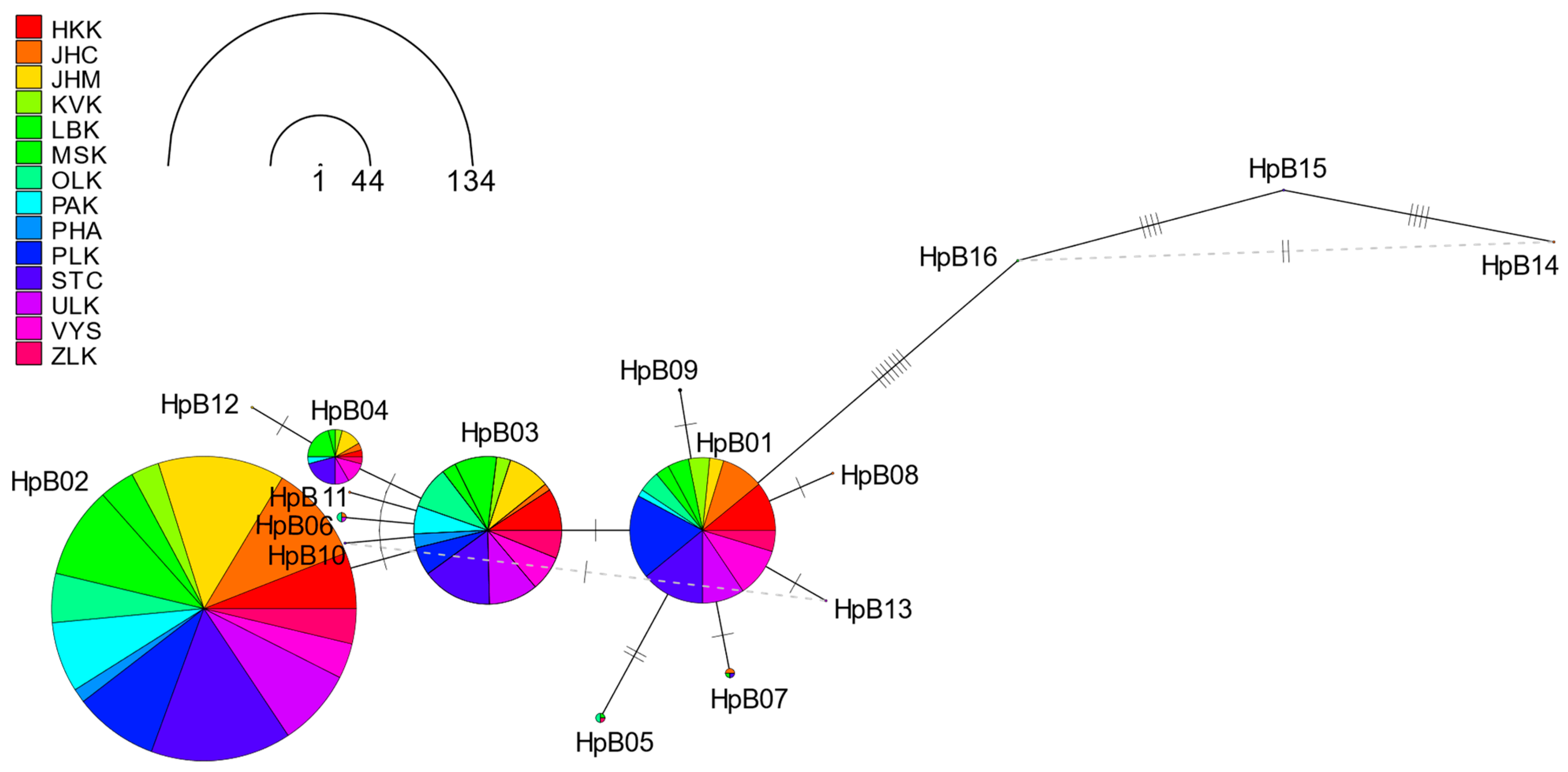
3.7. Haplotype Networks for tRNAleu-cox2 and cox1 Haplotypes and Regions in the Czech Republic
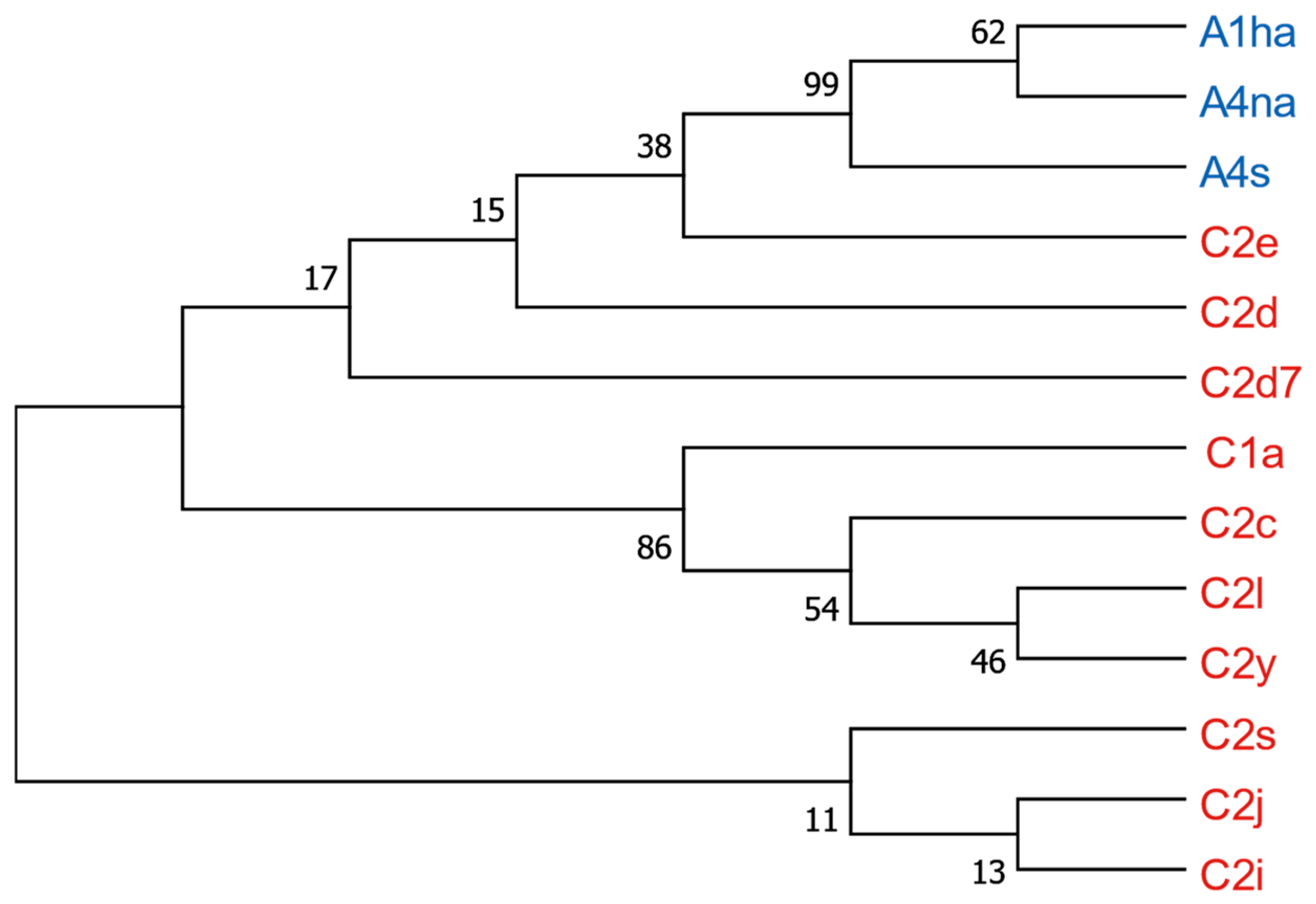
3.8. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitfield, C.W.; Behura, S.K.; Berlocher, S.H.; Clark, A.G.; Johnston, J.S.; Sheppard, W.S.; Smith, D.R.; Suarez, A.V.; Weaver, D.; Tsutsu, N.D. Thrice out of Africa: Ancient and recent expansions of the honey bee, Apis mellifera. Science 2006, 314, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Tihelka, E.; Cai, C.; Pisani, D.; Donoghue, P.C. Mitochondrial genomes illuminate the evolutionary history of the Western honey bee (Apis mellifera). Sci. Rep. 2020, 10, 14515. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Wallberg, A.; Webster, M.T. From where did the Western honeybee (Apis mellifera) originate? Ecol. Evol. 2012, 2, 1949–1957. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Z.; Pan, Q.; Chen, X.; Wang, H.; Guo, H.; Liu, S.; Lu, H.; Tian, S.; Li, R.; et al. Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n. ssp. Molec. Biol. Evol. 2016, 33, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Dogantzis, K.A.; Tiwari, T.; Conflitti, I.M.; Dey, A.; Patch, H.M.; Muli, E.M.; Garnery, L.; Whitfield, C.W.; Stolle, E.; Zayed, A. Thrice out of Asia and the adaptive radiation of the western honey bee. Sci. Adv. 2021, 7, eabj2151. [Google Scholar] [CrossRef] [PubMed]
- Franck, P.; Garnery, L.; Celebrano, G.; Solignac, M.; Cornuet, J.M. Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and Sicily (A. m. sicula). Molec. Ecol. 2000, 10, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Requier, F.; Garnery, L.; Kohl, P.L.; Njovu, H.K.; Pirk, C.W.W.; Crewe, R.M.; Steffan-Dewenter, I. The Conservation of Native Honey Bees Is Crucial. Trends. Ecol. Evol. 2019, 34, 789–798. [Google Scholar] [CrossRef]
- Bouga, M.; Harizanis, P.C.; Kilias, G.; Alahiotis, S. Genetic divergence and phylogenetic relationships of honey bee Apis mellifera (Hymenoptera: Apidae) populations from Greece and Cyprus using PCR–RFLP analysis of three mtDNA segments. Apidologie 2005, 36, 335–344. [Google Scholar] [CrossRef]
- Chávez-Galarza, J.; Henriques, D.; Johnston, J.S.; Carneiro, M.; Rufino, J.; Patton, J.C.; Pinto, M.A. Revisiting the Iberian honey bee (Apis mellifera iberiensis) contact zone: Maternal and genome-wide nuclear variations provide support for secondary contact from historical refugia. Molec. Ecol. 2015, 24, 2973–2992. [Google Scholar] [CrossRef]
- Bertrand, B.; Alburaki, M.; Legout, H.; Moulin, S.; Mougel, F.; Garnery, L. MtDNA COI-COII marker and drone congregation area: An efficient method to establish and monitor honeybee (Apis mellifera L.) conservation centres. Molec. Ecol Resour. 2015, 15, 673–683. [Google Scholar] [CrossRef]
- Meixner, M.D.; Costa, C.; Kryger, P.; Hatjina, F.; Bouga, M.; Ivanova, E.; Büchler, R. Conserving diversity and vitality for honey bee breeding. J. Apic. Res. 2010, 49, 85–92. [Google Scholar] [CrossRef]
- Pinto, M.A.; Henriques, D.; Chávez-Galarza, J.; Kryger, P.; Garnery, L.; van der Zee, R.; Dahle, B.; Soland-Reckeweg, G.; de la Rúa, P.; Dall’ Olio, R.; et al. Genetic integrity of the Dark European honey bee (Apis mellifera mellifera) from protected populations: A genome-wide assessment using SNPs and mtDNA sequence data. J. Apic. Res. 2014, 53, 269–278. [Google Scholar] [CrossRef]
- Büchler, R.; Costa, C.; Hatjina, F.; Andonov, S.; Meixner, M.D.; Le Conte, Y.; Uzunov, A.; Berg, S.; Bienkowska, M.; Bouga, M.; et al. The influence of genetic origin and its interaction with environmental effects on the survival of Apis mellifera L. colonies in Europe. J. Apic. Res. 2014, 53, 205–214. [Google Scholar] [CrossRef]
- Krejčík, P.; Scháňková, Š.; Mořický, J.; Chalupa, P. Situační a Výhledová Zpráva–Včely, 1st ed.; Ministerstvo Zemědělství: Prague, Czech Republic, 2021; p. 31. ISBN 978-80-7434-656-9. Available online: https://eagri.cz/public/portal/-q321807---Er5DAQ_w/situacni-a-vyhledova-zprava-vcely-2021?_linka=a552018 (accessed on 5 February 2024).
- van Engelsdorp, D.; Meixner, M.D. A historical review of managed honey bee populations in Europe and the United States and the factors that may affect them. J. Inverteb. Pathol. 2010, 103, S80–S95. [Google Scholar] [CrossRef]
- Texl, P.; Vondrák, J. Včely 2011—Co ministerstvo do zprávy nenapsalo. Moderní Včelař 2012, 9, 4–5. [Google Scholar]
- Goetze, G. Die beste Biene: Züchtungs-und Rassen-Kunde der Honigbiene nach dem Heutigen Stand von Wissenschaft und Praxis; Leipzig: Hamburg, Germany, 1940; pp. 64–198. [Google Scholar]
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin, Germany, 1988; p. 284. [Google Scholar]
- Tomšík, B. Apiar bioclimatical districts of Bohemia and Moravia and appreciation of the bee-family “Iskra II”. Acta Univ. Agricult. Silvicult. Brno Facultas Agricult. 1949, 30, 123. [Google Scholar]
- Tomšík, B. Včela středoevropská žije v Československu. Včelařství 1965, 18, 6–7. [Google Scholar]
- Veselý, V. Bewertung der importierten Rasse der Carnicabiene (Apis mellifera carnica Poll.) und der Hybriden derselben mit der hiesigen Biene in den Bedingungen der ČSR. Sci. Stud. Bee Res. Inst. Dol. 1976, 7, 137–157. [Google Scholar]
- Cori, E. O včele ušlechtilé a zušlechtění naší obyčejné včely. Český Včelař 1875, 9, 49–51, 61–65, 73–75, 85–89, 101–108, 137–139. Available online: https://dk.uzei.cz/uzei/periodical/uuid:06af2030-9848-418f-971e-c0eb5645c76a (accessed on 9 April 2024).
- Cornuet, J.M.; Garnery, L.; Solignac, M. Putative origin and function of the intergenic region between COI and COII of Apis mellifera L. mitochondrial DNA. Genetics 1991, 128, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Boardman, L.; Eimanifar, A.; Kimball, R.T.; Braun, E.L.; Fuchs, S.; Grünewald, B.; Ellis, J.D. The complete mitochondrial genome of Apis mellifera unicolor (Insecta: Hymenoptera: Apidae), the Malagasy honey bee. Mitochondrial DNA Part B 2019, 4, 3286–3287. [Google Scholar] [CrossRef]
- Chávez-Galarza, J.; Garnery, L.; Henriques, D.; Neves, C.J.; Loucif-Ayad, W.; Pinto, M.A. Mitochondrial DNA variation of Apis mellifera iberiensis: Further insights from a large-scale study using sequence data of the tRNAleu-cox2 intergenic region. Apidologie 2017, 48, 533–544. [Google Scholar] [CrossRef]
- Ajao, A.M.; Nneji, L.M.; Adeola, A.C.; Oladipo, S.O.; Ayoola, A.O.; Wang, Y.Y.; Adeniyi, A.V.; Olademeji, Y.U. Genetic diversity and population structure of the native Western African honeybee (Apis mellifera adansonii Latreille, 1804) in Nigeria based on mitochondrial COI sequences. Zool. Anz. 2021, 293, 17–25. [Google Scholar] [CrossRef]
- Cridland, J.M.; Tsutsui, N.D.; Ramírez, S.R. The complex demographic history and evolutionary origin of the western honey bee, Apis mellifera. Genome Biol. Evol. 2017, 9, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Cleary, D.; Szalanski, A.L.; Trammel, C.; Williams, M.K.; Tripodi, A.; Downey, D. Mitochondrial DNA Variation of Feral Honey Bees (L.) from Utah (USA). J. Apic. Sci. 2018, 62, 223–232. [Google Scholar] [CrossRef]
- Oleksa, A.; Kusza, S.; Tofilski, A. Mitochondrial DNA Suggests the Introduction of Honeybees of African Ancestry to East-Central Europe. Insects 2021, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Alburaki, M.; Madella, S.; Lopez, J.; Bouga, M.; Chen, Y.; vanEngelsdorp, D. Honey bee populations of the USA display restrictions in their mtDNA haplotype diversity. Front. Gen. 2023, 13, 3566. [Google Scholar] [CrossRef]
- Sušnik, S.; Kozmus, P.; Poklukar, J.; Meglic, V. Molecular characterisation of indigenous Apis mellifera carnica in Slovenia. Apidologie 2004, 35, 623–636. [Google Scholar] [CrossRef]
- Muñoz, I.; Dall’Olio, R.; Lodesani, M.; de La Rúa, P. Population genetic structure of coastal Croatian honeybees (Apis mellifera carnica). Apidologie 2009, 40, 617–626. [Google Scholar] [CrossRef]
- Péntek-Zakar, E.; Oleksa, A.; Borowik, T.; Kusza, S. Population structure of honey bees in the Carpathian Basin (Hungary) confirms introgression from surrounding subspecies. Ecol. Evol. 2015, 5, 5456–5467. [Google Scholar] [CrossRef] [PubMed]
- Kaskinova, M.D.; Gaifullina, L.R.; Saltykova, E.S. Haplotypes of the tRNAleu-COII mtDNA Region in Russian Apis mellifera Populations. Animals 2023, 13, 2394. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Ratnasingham, S.; de Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. B 2003, 270, S96–S99. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D. The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Garnery, L. Mitochondrial DNA variability in honeybees and its phylogeographic implications. Apidologie 1991, 22, 627–642. [Google Scholar] [CrossRef]
- Garnery, L.; Cornuet, J.M.; Solignac, M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1992, 1, 145–154. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Garnery, L.; Solignac, M.; Celebrano, G.; Cornuet, J.-M. A simple test using restricted PCR-amplified mitochondrial DNA to study the genetic structure of Apis mellifera L. Experientia 1993, 49, 1016–1021. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Nei, M.; Tajima, F. DNA polymorphism detectable by restriction endonuclease. Genetics 1981, 97, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 595. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E. Analysis of haplotype networks: The randomized minimum spanning tree method. Methods Ecol. Evol. 2018, 9, 1308–1317. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 15 December 2023).
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Pagač, M. K dovozu kraňky do českých zemí. Včelařství 1990, 2, 31. [Google Scholar]
- Texl, P.; Přidal, A.; Rytina, L.; Holub, P.; Klíma, Z.; Gruna, B.; Matela, L.; Kala, J.; Jůzek, M.; Čížková, P. Na stopě původní včely v šumavských hvozdech. (Traces of the indigenous honeybee population in Šumava). Moderní Včelař 2010, 7, 116–118. [Google Scholar]
- Texl, P.; Přidal, A. Biodiverzita a hledání tmavé včely na Šumavě. (Biodiversity and looking for the Dark bee in Šumava). Šumava 2010, 18–19. [Google Scholar]
- Fencl, J. Plemenné Vyhodnocení Naší Včely (Race Evaluation of the Czech Honeybee). Master’s Thesis, Agriculture University in Brno, Brno, Czech Republic, 1964. [Google Scholar]
- Jílek, J. Morfologický a Fysiologický Rozbor včel Kmene Moravského a Drahanské Vysočiny. (Morphological and Fysiological Evaluation of the Moravian Honeybee Strain from Drahanská Highlands). Master’s Thesis, Agriculture University in Brno, Brno, Czech Republic, 1962. [Google Scholar]
- Tanasković, M.; Erić, P.; Patenković, A.; Erić, K.; Mihajlović, M.; Tanasić, V.; Stanisavljević, L.; Davidović, S. MtDNA Analysis Indicates Human-Induced Temporal Changes of Serbian Honey Bees Diversity. Insects 2021, 12, 767. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.; De la Rúa, P. Wide genetic diversity in Old World honey bees threaten by introgression. Apidologie 2021, 52, 200–217. [Google Scholar] [CrossRef]
- Szalanski, A.L.; Magnus, R.M. Mitochondrial DNA characterization of Africanized honey bee (Apis mellifera L.) populations from the USA. J. Apic. Res. 2010, 49, 177–185. [Google Scholar] [CrossRef]
- Osterlund, E. The Elgon Bee—A Hobbyist and Commercial Bee with African Genes. Am. Bee J. 1993, 133, 504–507. [Google Scholar]
- Živanský, F. Milí ctění členové spolkoví! Včela Brněnská 1872, 6, 1–5. [Google Scholar]
- Veselý, V. Strain crossing on mating stations and evaluation of further hybridisation by means of artificial insemination. Sci. Stud. Bee Res. Inst. Dol. 1968, 5, 141–173. [Google Scholar]
- Triseleva, T.A.; Safonkin, A.F.; Bykova, T.O.; Rukhkyan, M.J. Intrabreed Diversity and Relationships between Races of the Honey Bee Apis mellifera carpathica and Apis mellifera caucasica. Biol. Bull. Russ. Acad. Sci. 2023, 50, 546–554. [Google Scholar] [CrossRef]
- Syromyatnikov, M.Y.; Borodachev, A.V.; Kokina, A.V.; Popov, V.N. A Molecular Method for the Identification of Honey Bee Subspecies Used by Beekeepers in Russia. Insects 2018, 9, 10. [Google Scholar] [CrossRef]
- Henriques, D.; Chávez-Galarza, J.; Quaresma, A.; Neves, C.J.; Lopes, R.A.; Costa, C.; Costa, F.O.; Rufino, J.; Pinto, M.A. From the popular tRNAleu-COX2 intergenic region to the mitogenome: Insights from diverse honey bee populations of Europe and North Africa. Apidologie 2019, 50, 215–229. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L | G | Haplotype | NCBI Accession | DraI Spectrum (bp) | Seq. Length (bp) | NCBI Reference | |
|---|---|---|---|---|---|---|---|
| Accession Number (% Similarity) | Designated as | ||||||
| C | Q | C1a | PP350804 | 47, 41, 64, 420 | 572 | JQ977699.1 (100) | carnica |
| C | Q | C2l | PP350810 | 47, 40, 64, 419 | 570 | OR761864.1 (100) | carnica |
| C | Q | C2e | PP350812 | 47, 40, 63, 420 | 570 | JQ977702.1 (100) | carnica |
| C | Q | C2d | PP350805 | 47, 40, 64, 420 | 571 | JF723977.1 (100) | carnica |
| C | Q | C2c | PP350806 | 47, 40, 64, 420 | 571 | JF723976.1 (100) | carnica |
| C | Q | C2j | PP350807 | 47, 40, 64, 420 | 571 | KX463941.1 (100) | carnica |
| C | Q | C2s | PP350808 | 47, 40, 63, 421 | 571 | JF723979.1 (100) | carnica |
| C | Q | C2d7 * | PP397042 | 46, 40, 64, 420 | 570 | JF723977.1 (99.81) | carnica |
| C | Q | C2i | PP350809 | 47, 40, 64, 420 | 571 | JQ977703.1 (100) | carnica |
| C | Q | C2y | PP350811 | 47, 40, 64, 420 | 571 | JQ754650.1 (100) | carnica |
| A | P0Q | A1ha * | PP430326 | 47, 108, 482 | 637 | KX463739.1 (99.83) | iberiensis |
| A | P0QQ | A4s | PP430327 | 47, 107, 192, 483 | 829 | MW939597.1 (100) | n.a. |
| A | P0QQ | A4na * | PP430328 | 47, 108, 190, 482 | 827 | KX463793.1 (99.75) | iberiensis |
| Haplotype | Sampled in Hives | Sampled on Flowers | Total | |||
|---|---|---|---|---|---|---|
| N | Frequency | N | Frequency | N | Frequency | |
| C1a | 72 | 0.4675 | 83 | 0.5390 | 155 | 0.5032 |
| C2l | 15 | 0.0974 | 10 | 0.0649 | 25 | 0.0812 |
| C2e | 34 | 0.2208 | 24 | 0.1558 | 58 | 0.1883 |
| C2d | 7 | 0.0455 | 5 | 0.0325 | 12 | 0.0390 |
| C2c | 19 | 0.1234 | 28 | 0.1818 | 47 | 0.1526 |
| C2j | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| C2s | 4 | 0.0260 | 0 | 0.0000 | 4 | 0.0130 |
| C2d7 | 0 | 0 | 1 | 0.0065 | 1 | 0.0032 |
| C2i | 0 | 0 | 1 | 0.0065 | 1 | 0.0032 |
| C2y | 0 | 0 | 1 | 0.0065 | 1 | 0.0032 |
| A1ha | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| A4s | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| A4na | 0 | 0 | 1 | 0.0065 | 1 | 0.0032 |
| Total | 154 | 1.0000 | 154 | 1.0000 | 308 | 1.0000 |
| Haplotype | Sampled in Hives | Sampled on Flowers | Total | |||
|---|---|---|---|---|---|---|
| N | Frequency | N | Frequency | N | Frequency | |
| HpB01 | 37 | 0.2403 | 27 | 0.1753 | 64 | 0.2078 |
| HpB02 | 68 | 0.4416 | 66 | 0.4286 | 134 | 0.4351 |
| HpB03 | 24 | 0.1558 | 41 | 0.2662 | 65 | 0.2110 |
| HpB04 | 14 | 0.0909 | 10 | 0.0649 | 24 | 0.0779 |
| HpB05 | 3 | 0.0195 | 1 | 0.0065 | 4 | 0.0130 |
| HpB06 | 0 | 0.0000 | 4 | 0.0260 | 4 | 0.0130 |
| HpB07 | 3 | 0.0195 | 1 | 0.0065 | 4 | 0.0130 |
| HpB08 | 0 | 0.0000 | 1 | 0.0065 | 1 | 0.0032 |
| HpB09 | 0 | 0.0000 | 1 | 0.0065 | 1 | 0.0032 |
| HpB10 | 0 | 0.0000 | 1 | 0.0065 | 1 | 0.0032 |
| HpB11 | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| HpB12 | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| HpB13 | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| HpB14 | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| HpB15 | 1 | 0.0065 | 0 | 0.0000 | 1 | 0.0032 |
| HpB16 | 0 | 0.0000 | 1 | 0.0065 | 1 | 0.0032 |
| Total | 154 | 1.0000 | 154 | 1.0000 | 308 | 1.0000 |
| Haplotype Sampling | N | H | Hd | π | D |
|---|---|---|---|---|---|
| tRNAleu-cox2 (796 bp) | |||||
| In hives | 154 | 9 | 0.70970 | 0.00201 (1.7583 × 10−6) | −3.17613 (0.00149) |
| On flowers | 154 | 9 | 0.65096 | 0.00139 (1.02984 × 10−6) | −3.19736 (0.00139) |
| Total | 308 | 13 | 0.68186 | 0.00172 (1.38726 × 10−6) | −3.02386 (0.00249) |
| cox1 (658 bp) | |||||
| In hives | 154 | 11 | 0.71845 | 0.00229 (2.35701 × 10−6) | −1.58841 (0.11219) |
| On flowers | 154 | 11 | 0.71420 | 0.00177 (1.61582 × 10−6) | −1.56860 (0.11674) |
| Total | 308 | 16 | 0.71866 | 0.00203 (1.96124 × 10−6) | −1.60658 (0.10815) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knoll, A.; Langová, L.; Přidal, A.; Urban, T. Haplotype Diversity in mtDNA of Honeybee in the Czech Republic Confirms Complete Replacement of Autochthonous Population with the C Lineage. Insects 2024, 15, 495. https://doi.org/10.3390/insects15070495
Knoll A, Langová L, Přidal A, Urban T. Haplotype Diversity in mtDNA of Honeybee in the Czech Republic Confirms Complete Replacement of Autochthonous Population with the C Lineage. Insects. 2024; 15(7):495. https://doi.org/10.3390/insects15070495
Chicago/Turabian StyleKnoll, Aleš, Lucie Langová, Antonín Přidal, and Tomáš Urban. 2024. "Haplotype Diversity in mtDNA of Honeybee in the Czech Republic Confirms Complete Replacement of Autochthonous Population with the C Lineage" Insects 15, no. 7: 495. https://doi.org/10.3390/insects15070495