
Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae)
 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Sequencing
2.2. Assembly, Annotation, and Composition Analyses
2.3. Phylogenetic Relationship
3. Results and Discussions
3.1. Mitogenomic Structure
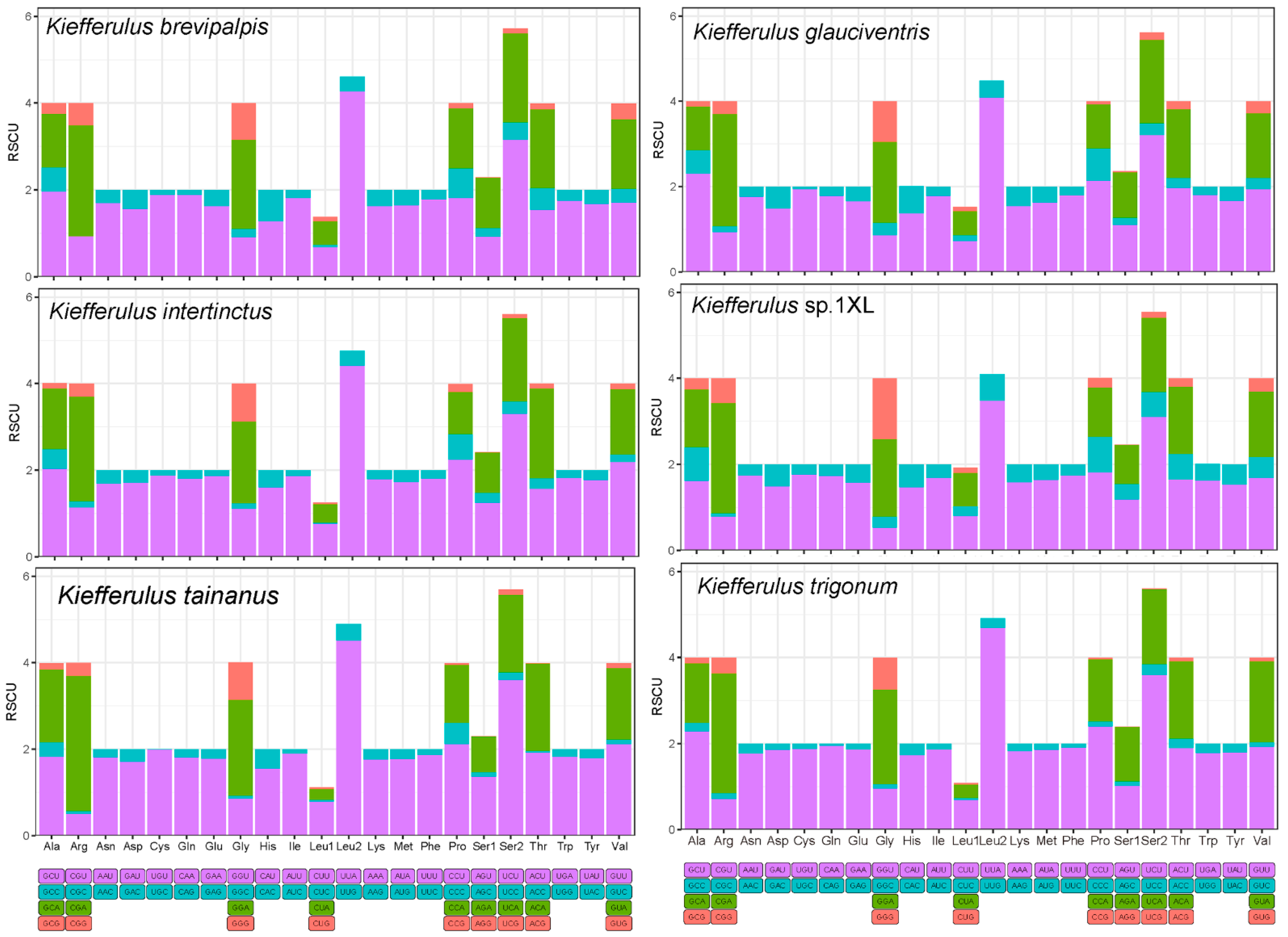
3.2. Protein-Coding Genes, Codon Usage, and Evolutionary Rates
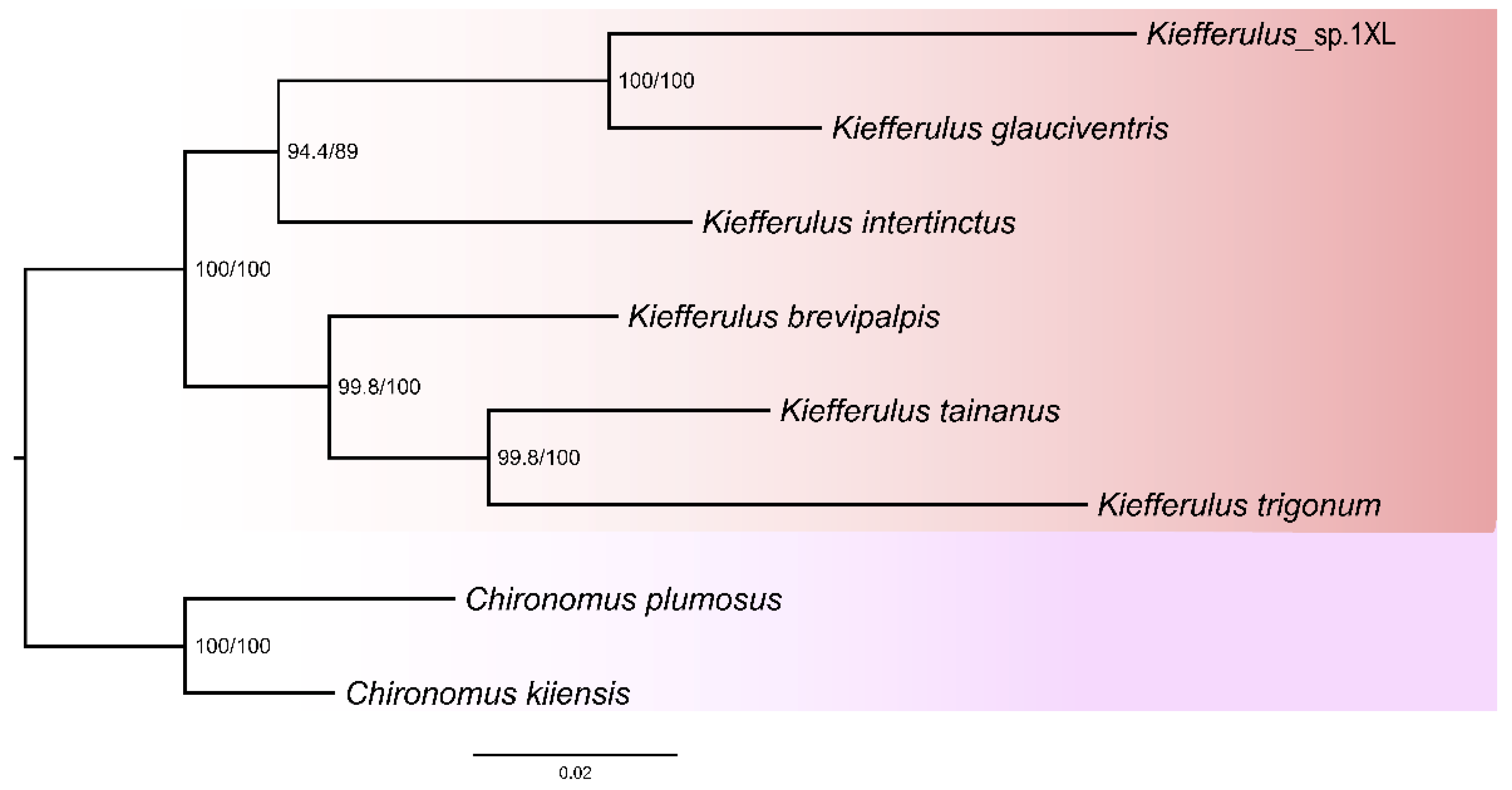
3.3. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lencioni, V.; Cranston, P.S.; Makarchenko, E.J. Recent advances in the study of Chironomidae: An overview. J. Limnol. 2018, 77, 1–6. [Google Scholar] [CrossRef]
- Lin, X.L.; Zhao, Y.M.; Yan, L.P.; Liu, W.B.; Bu, W.J.; Wang, X.H.; Zheng, C.G. Mitogenomes provide new insights into the evolutionary history of Prodiamesinae (Diptera: Chironomidae). Zool. Scr. 2022, 51, 119–132. [Google Scholar] [CrossRef]
- Lin, X.L.; Liu, Z.; Yan, L.P.; Duan, X.; Bu, W.J.; Wang, X.H.; Zheng, C.G. Mitogenomes provide new insights of evolutionary history of Boreheptagyiini and Diamesini (Diptera: Chironomidae: Diamesinae). Ecol. Evol. 2022, 12, e8957. [Google Scholar] [CrossRef]
- Armitage, P.D.; Pinder, L.; Cranston, P. The Chironomidae: Biology and Ecology of Non-Biting Midges; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Giłka, W.; Zakrzewska, M.; Lukashevich, E.D.; Vorontsov, D.D.; Soszyńska-Maj, A.; Skibińska, K.; Cranston, P.S. Wanted, tracked down and identified: Mesozoic non-biting midges of the subfamily Chironominae (Chironomidae, Diptera). Zool. J. Linn. Soc-Lond. 2021, 194, 874–892. [Google Scholar] [CrossRef]
- Song, C.; Wang, X.; Bu, W.; Qi, X. Morphology lies: A case-in-point with a new non-biting midge species from Oriental China (Diptera, Chironomidae). Zookeys 2020, 909, 67. [Google Scholar] [CrossRef]
- Epler, J.; Ekrem, T.; Cranston, P.J.; Cranston, P.S.; Epler, J. The larvae of Chironomidae of the holarctic region-keys and diagnoses. Insect Syst. Evol. 2013, 66, 387–556. [Google Scholar] [CrossRef]
- Cranston, P.S. The Chironomidae larvae associated with the tsunami-impacted waterbodies of the coastal plain of southwestern Thailand. Bull. Raffles Mus. 2007, 55, 231–244. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Peng, L.; Vogler, A.P.; Morse, J.C.; Yang, L.; Sun, C.; Wang, B. Massive gene rearrangements of mitochondrial genomes and implications for the phylogeny of Trichoptera (Insecta). Syst. Entomol. 2022, 48, 278–295. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Brown, W.M. The mitochondrial genome of animals. In Molecular Evolutionary Genetics; Plenum Press: New York, NY, USA, 1985; pp. 95–130. [Google Scholar]
- Li, X.; Yan, L.; Pape, T.; Gao, Y.; Zhang, D. Evolutionary insights into bot flies (Insecta: Diptera: Oestridae) from comparative analysis of the mitochondrial genomes. Int. J. Biol. Macromol. 2020, 149, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yan, L.; Gao, Y.; Zhang, D. First report of mitochondrial genome from the subfamily Bengaliinae (Diptera: Calliphoridae). Mitochondrial DNA Part B 2019, 4, 1560–1561. [Google Scholar] [CrossRef]
- Zhang, D.; He, F.X.; Li, X.B.; Aishan, Z.; Lin, X.L. New mitogenomes of the Polypedilum generic complex (Diptera: Chironomidae): Characterization and phylogenetic implications. Insects 2023, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, X.; Mao, B.; Xiao, Y.; Shen, M.; Fu, Y. Comparative mitogenome analyses of twelve non-biting flies and provide insights into the phylogeny of Chironomidae (Diptera: Culicomorpha). Sci. Rep. 2023, 13, 9200. [Google Scholar] [CrossRef]
- Cranston, P.S.; Webb, C.J.; Martin, J.J. The saline nuisance chironomid Carteronica longilobus (Diptera: Chironomidae): A systematic reappraisal. Syst. Entomol. 1990, 15, 401–432. [Google Scholar] [CrossRef]
- Li, S.Y.; Zhao, Y.M.; Guo, B.X.; Li, C.H.; Sun, B.J.; Lin, X.L. Comparative analysis of mitogenomes of Chironomus (Diptera: Chironomidae). Insects 2022, 13, 1164. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G.J. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.-M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F.J. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T.J.B. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Longo, G.C. FASconCAT-G: Extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 2014, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Wang, H.C.; Minh, B.Q.; Susko, E.; Roger, A.J. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst. Biol. 2017, 67, 216–235. [Google Scholar] [CrossRef]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J.J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.G.; Liu, Z.; Zhao, Y.M.; Wang, Y.; Bu, W.J.; Wang, X.H.; Lin, X.L. First report on mitochondrial gene rearrangement in non-biting midges, revealing a synapomorphy in Stenochironomus Kieffer (Diptera: Chironomidae). Insects 2022, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Jo, H.; Choi, B.; Kwak, I.S. Complete mitochondrial genome of Stictochironomus akizukii (Tokunaga)(Chironomidae, Diptera) assembled from next-generation sequencing data. Mitochondrial DNA Part B 2020, 5, 2310–2311. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zang, H.; Ye, X.; Peng, L.; Wang, B.; Lian, G.; Sun, C.J. Comparative mitogenomic analyses of hydropsychidae revealing the novel rearrangement of protein-coding gene and tRNA (Trichoptera: Annulipalpia). Insects 2022, 13, 759. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Service, M. Female genitalia in Chironomidae and other Nematocera: Morphology, phylogenies, keys. Bull. Fish. Res. Board Can. 1977, 197, 1–209. [Google Scholar]
- Martin, J.; Blinov, A.; Alieva, E.; Hirabayashi, K. A molecular phylogenetic investigation of the genera closely related to Chironomus Meigen (Diptera: Chironomidae). In Contributions to the Systematics and Ecology of Aquatic Diptera. A Tribute to Ole A. Sæther; Andersen, T., Ed.; Caddis Press: Columbus, OH, USA, 2007; pp. 193–203. [Google Scholar]
- Tang, H.; Cheng, Q.; Krosch, M.N.; Cranston, P.S. Maritime midge radiations in the Pacific Ocean (Diptera: Chironomidae). Syst. Entomol. 2023, 48, 111–126. [Google Scholar] [CrossRef]
- Zhang, D.; Niu, Z.Q.; Luo, A.R.; Orr, M.C.; Ferrari, R.R.; Jin, J.F.; Wu, Q.T.; Zhang, F.; Zhu, C.D. Testing the systematic status of Homalictus and Rostrohalictus with weakened cross-vein groups within Halictini (Hymenoptera: Halictidae) using low-coverage whole-genome sequencing. Insect Sci. 2022, 29, 1819–1833. [Google Scholar] [CrossRef]
- Zhang, F.; Ding, Y.; Zhu, C.D.; Zhou, X.; Orr, M.C.; Scheu, S.; Luan, Y.X. Phylogenomics from low-coverage whole-genome sequencing. Methods Ecol. Evol. 2019, 10, 507–517. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Location | Longitude and Latitude | Elevation (m) | Life Stage | Date | Collector | Accession Number | References |
|---|---|---|---|---|---|---|---|---|
| Kiefferulus sp.1XL | Namibia, Otjozondjupa | 16.745° E, −22.111° N | 1220 | Adult | 4 December 2018 | Xiaolong Lin | PP884096 | This study |
| Kiefferulus brevipalpis | Namibia, Otjozondjupa | 16.745° E, −22.111° N | 1220 | Adult | 4 December 2018 | Xiaolong Lin | PP884099 | This study |
| Kiefferulus glauciventris | China, Xishuangbanna | 99.989° E, 22.055° N | 700 | Adult | 6 May 2013 | Xiaolong Lin | PP884098 | This study |
| Kiefferulus intertinctus | New Caledonia, Tontouta | 166.206° E, −22.008° N | 10 | Pupa | 11 May 2018 | Nathalie Mary | PP884097 | This study |
| Kiefferulus tainanus | China, Guangzhou | 113.959° E, 23.339° N | 149 | Adult | 12 April 2018 | Xiaolong Lin | PP972215 | This study |
| Kiefferulus trigonum | China, Huanggang | 115.738° E, 31.095° N | 552 | Adult | 11 July 2021 | Shuang Qiu | PP884095 | This study |
| Species | Regions | Length (bp) | A | T | C | G | AT | GC | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|---|
| Kiefferulus brevipalpis | Whole genome | 15,767 | 40.21 | 37.12 | 13.64 | 9.02 | 77.33 | 22.66 | 0.040 | −0.204 |
| PCGs | 11,619 | 31.22 | 44.50 | 12.85 | 11.43 | 75.72 | 24.27 | −0.175 | −0.059 | |
| Site 1 | 4141 | 31.93 | 38.64 | 12.07 | 17.35 | 70.57 | 29.43 | −0.090 | 0.155 | |
| Site 2 | 3739 | 21.47 | 48.55 | 18.01 | 11.95 | 70.02 | 29.96 | −0.385 | −0.203 | |
| Site 3 | 3739 | 40.26 | 46.30 | 8.46 | 4.98 | 86.56 | 13.44 | −0.069 | −0.233 | |
| tRNA | 1488 | 41.06 | 38.64 | 8.67 | 11.63 | 79.70 | 20.30 | 0.030 | 0.146 | |
| l-rRNA | 1381 | 41.20 | 43.95 | 4.71 | 10.14 | 85.16 | 14.84 | −0.032 | 0.366 | |
| s-rRNA | 815 | 42.82 | 40.61 | 5.40 | 11.17 | 83.44 | 16.56 | 0.026 | 0.348 | |
| CR | 674 | 46.44 | 49.11 | 2.67 | 1.78 | 95.55 | 4.45 | −0.028 | −0.200 | |
| Kiefferulus glauciventris | Whole genome | 15,644 | 39.24 | 37.77 | 13.84 | 9.15 | 77.01 | 22.99 | 0.019 | −0.204 |
| PCGs | 11,619 | 30.48 | 44.62 | 13.10 | 11.80 | 75.10 | 24.90 | −0.188 | −0.052 | |
| Site 1 | 4141 | 31.41 | 37.34 | 13.13 | 18.12 | 68.75 | 31.25 | −0.080 | 0.161 | |
| Site 2 | 3739 | 21.45 | 48.57 | 18.04 | 11.93 | 70.02 | 29.98 | −0.385 | −0.206 | |
| Site 3 | 3739 | 38.59 | 47.94 | 8.12 | 5.34 | 86.53 | 13.47 | −0.108 | −0.200 | |
| tRNA | 1487 | 41.29 | 38.53 | 8.88 | 11.30 | 79.83 | 20.17 | 0.035 | 0.120 | |
| l-rRNA | 1368 | 43.64 | 41.08 | 4.68 | 10.60 | 84.72 | 15.28 | 0.030 | 0.388 | |
| s-rRNA | 810 | 42.10 | 41.23 | 5.43 | 11.23 | 83.33 | 16.67 | 0.010 | 0.348 | |
| CR | 568 | 49.47 | 45.77 | 2.64 | 2.11 | 95.25 | 4.75 | 0.039 | −0.111 | |
| Kiefferulus intertinctus | Whole genome | 15,644 | 39.87 | 38.24 | 13.05 | 8.84 | 78.11 | 21.89 | 0.021 | −0.192 |
| PCGs | 11,619 | 31.13 | 45.34 | 12.33 | 11.20 | 76.47 | 23.53 | −0.186 | −0.048 | |
| Site 1 | 4141 | 31.76 | 38.38 | 12.10 | 17.75 | 70.14 | 29.86 | −0.088 | 0.185 | |
| Site 2 | 3739 | 21.48 | 48.09 | 18.44 | 11.98 | 69.58 | 30.42 | −0.379 | −0.213 | |
| Site 3 | 3739 | 40.16 | 49.54 | 6.44 | 3.86 | 89.70 | 10.30 | −0.104 | −0.235 | |
| Kiefferulus intertinctus | tRNA | 1490 | 41.07 | 39.13 | 8.46 | 11.34 | 80.20 | 19.80 | 0.024 | 0.146 |
| l-rRNA | 1376 | 42.08 | 43.31 | 4.65 | 9.96 | 85.39 | 14.61 | −0.014 | 0.363 | |
| s-rRNA | 815 | 42.58 | 41.23 | 5.40 | 10.80 | 83.80 | 16.20 | 0.016 | 0.333 | |
| CR | 533 | 43.71 | 51.78 | 1.88 | 2.63 | 95.50 | 4.50 | −0.085 | 0.167 | |
| Kiefferulus sp.1XL | Whole genome | 15,588 | 38.42 | 36.70 | 15.03 | 9.85 | 75.12 | 24.88 | 0.023 | −0.208 |
| PCGs | 11,619 | 29.94 | 43.16 | 14.58 | 12.32 | 73.10 | 26.90 | −0.181 | −0.084 | |
| Site 1 | 4141 | 31.84 | 36.34 | 13.80 | 18.02 | 68.18 | 31.82 | −0.062 | 0.115 | |
| Site 2 | 3739 | 21.19 | 48.13 | 18.61 | 12.06 | 69.33 | 30.67 | −0.385 | −0.214 | |
| Site 3 | 3739 | 36.79 | 45.00 | 11.32 | 6.89 | 81.79 | 18.21 | −0.100 | −0.256 | |
| tRNA | 1488 | 40.39 | 38.98 | 9.07 | 11.56 | 79.37 | 20.63 | 0.018 | 0.121 | |
| l-rRNA | 1369 | 43.83 | 40.32 | 5.26 | 10.59 | 84.15 | 15.85 | 0.042 | 0.336 | |
| s-rRNA | 811 | 40.57 | 41.43 | 5.67 | 12.33 | 82.00 | 18.00 | −0.010 | 0.370 | |
| CR | 546 | 48.17 | 46.34 | 3.48 | 2.01 | 94.51 | 5.49 | 0.019 | −0.267 | |
| Kiefferulus trigonum | Whole genome | 15,657 | 40.47 | 38.97 | 12.19 | 8.37 | 79.44 | 20.56 | 0.019 | −0.186 |
| PCGs | 11,621 | 31.82 | 46.42 | 11.21 | 10.54 | 78.24 | 21.76 | −0.187 | −0.031 | |
| Site 1 | 4141 | 32.52 | 39.19 | 11.40 | 16.88 | 71.72 | 28.28 | −0.090 | 0.163 | |
| Site 2 | 3740 | 21.28 | 49.01 | 17.84 | 11.87 | 70.29 | 29.71 | −0.392 | −0.205 | |
| Site 3 | 3740 | 41.67 | 51.05 | 4.40 | 2.87 | 92.72 | 7.28 | −0.101 | −0.180 | |
| tRNA | 1484 | 41.98 | 38.88 | 8.09 | 11.05 | 80.86 | 19.14 | 0.038 | 0.155 | |
| l-rRNA | 1378 | 41.36 | 43.90 | 4.57 | 10.16 | 85.27 | 14.73 | −0.030 | 0.379 | |
| s-rRNA | 812 | 44.09 | 40.76 | 5.30 | 9.85 | 84.85 | 15.15 | 0.039 | 0.301 | |
| CR | 518 | 44.98 | 51.16 | 2.70 | 1.16 | 96.14 | 3.86 | −0.064 | −0.400 | |
| Kiefferulus tainanus | Whole genome | 15,748 | 40.15 | 38.67 | 12.56 | 8.62 | 78.82 | 21.18 | 0.018 | −18.620 |
| PCGs | 11,220 | 30.77 | 45.41 | 11.75 | 12.08 | 76.18 | 23.82 | −0.192 | 1.380 | |
| Site 1 | 3740 | 31.34 | 38.05 | 11.9 | 18.72 | 69.39 | 30.61 | −0.096 | 22.27 | |
| Site 2 | 3740 | 20.43 | 47.51 | 18.96 | 13.10 | 67.94 | 32.06 | −0.398 | −18.27 | |
| Site 3 | 3740 | 40.53 | 50.67 | 4.39 | 4.41 | 91.20 | 8.80 | −0.111 | 0.300 | |
| tRNA | 1494 | 41.5 | 38.76 | 8.43 | 11.31 | 80.25 | 19.75 | 0.034 | 14.580 | |
| l-rRNA | 1388 | 43.23 | 42.51 | 4.54 | 9.73 | 85.73 | 14.27 | 0.008 | 36.360 | |
| s-rRNA | 814 | 43.24 | 41.28 | 5.16 | 10.32 | 84.52 | 15.48 | 0.023 | 33.330 | |
| CR | 554 | 45.67 | 49.28 | 3.43 | 1.62 | 94.95 | 5.05 | −0.038 | −35.710 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Jin, W.-D.; Xu, H.-F.; Li, X.-B.; Jiang, Y.-W.; Li, D.-Q.; Lin, X.-L. Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae). Insects 2024, 15, 646. https://doi.org/10.3390/insects15090646
Zhang D, Jin W-D, Xu H-F, Li X-B, Jiang Y-W, Li D-Q, Lin X-L. Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae). Insects. 2024; 15(9):646. https://doi.org/10.3390/insects15090646
Chicago/Turabian StyleZhang, Dan, Wei-Dong Jin, Hai-Feng Xu, Xue-Bo Li, Yong-Wei Jiang, Dai-Qing Li, and Xiao-Long Lin. 2024. "Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae)" Insects 15, no. 9: 646. https://doi.org/10.3390/insects15090646
APA StyleZhang, D., Jin, W.-D., Xu, H.-F., Li, X.-B., Jiang, Y.-W., Li, D.-Q., & Lin, X.-L. (2024). Characteristics and Comparative Analysis of Six Mitogenomes of Genus Kiefferulus Goetghebuer, 1922 (Diptera: Chironomidae). Insects, 15(9), 646. https://doi.org/10.3390/insects15090646