
Evolutionary Relationships of Omani Macrotermes subhyalinus, Macrotermitinae

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Sampling
2.2. DNA Extraction
2.2.1. COI
2.2.2. 28S
2.3. Amplification and Sequencing Protocol
2.4. Phylogenetic Analysis
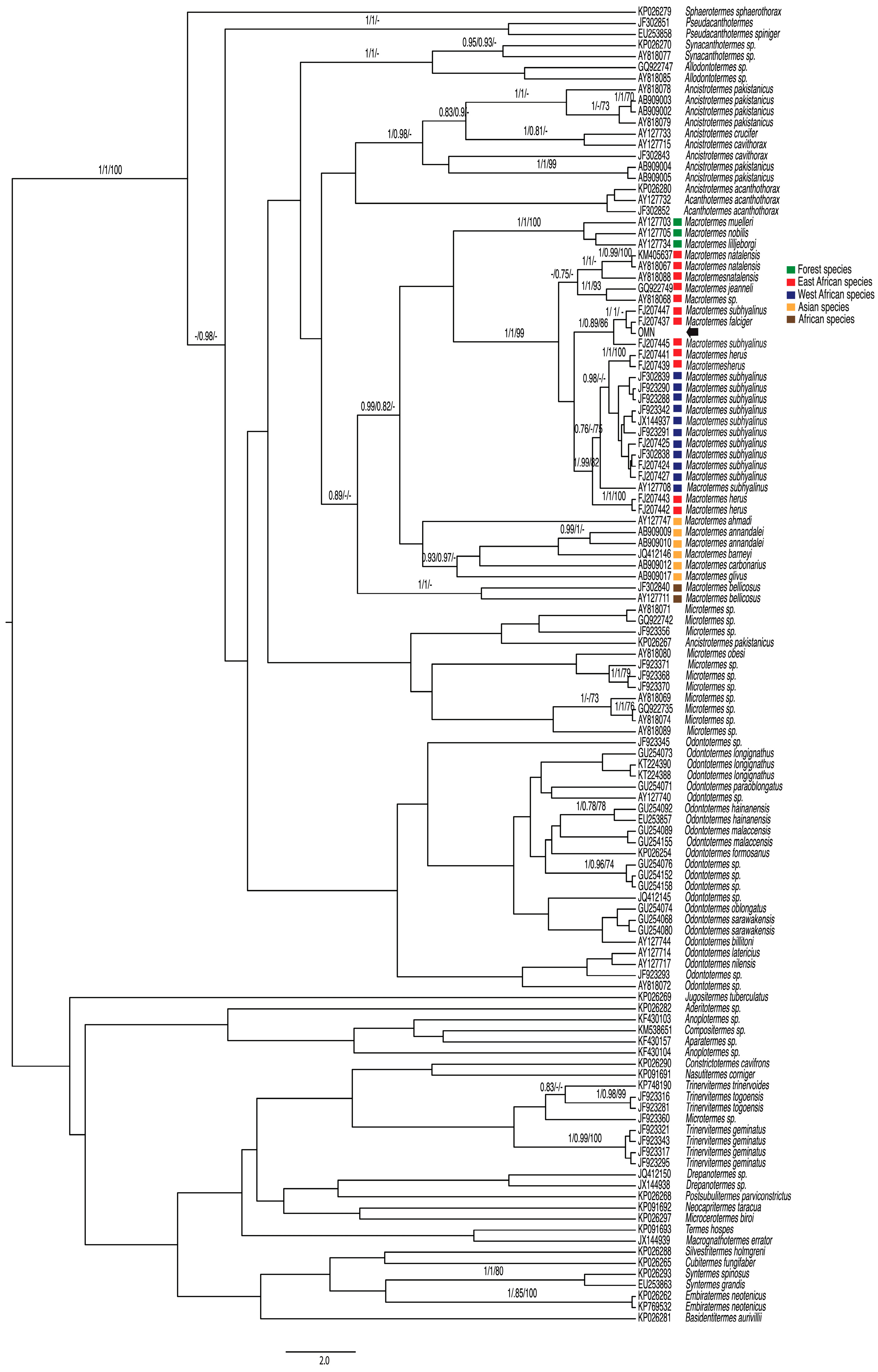
3. Results
3.1. COI
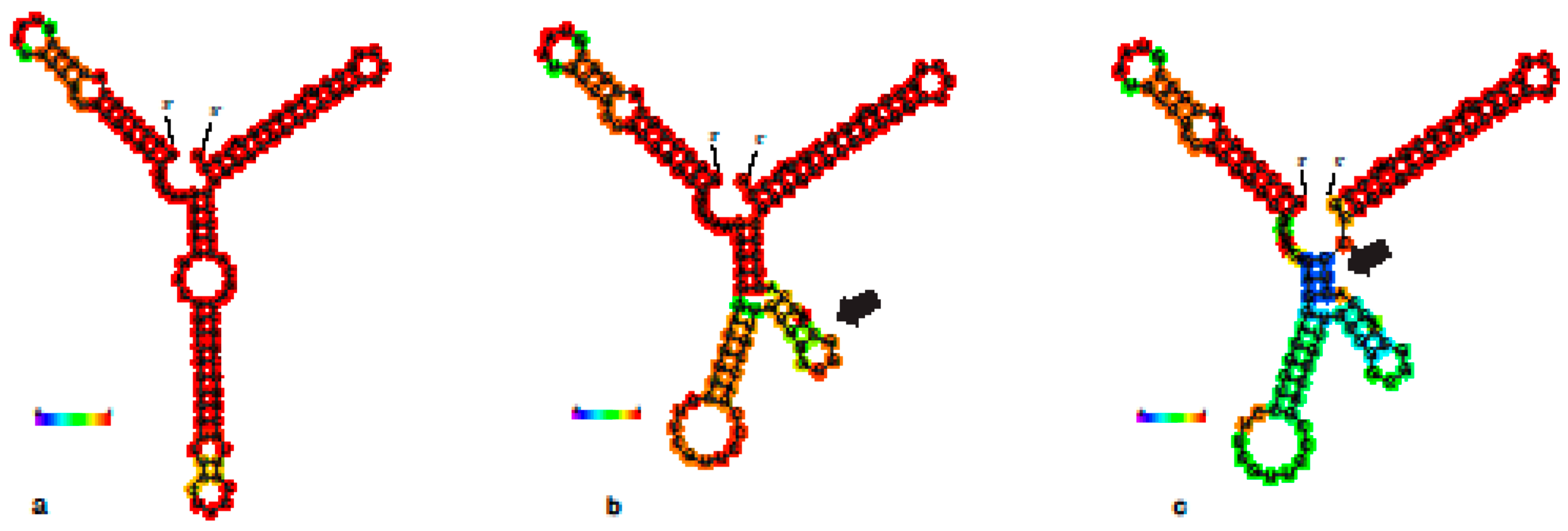
3.2. 28S
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rouland-Lefèvre, C. Symbiosis with fungi. In Termites: Evolution, Sociality, Symbioses, Ecology; Springer: Berlin/Heidelberg, Germany, 2000; pp. 289–306. [Google Scholar]
- Sieber, R. Establishment of fungus comb in Laboratory colonies ofMacrotermes michaelseni andOdontotermes montanus (Isoptera, Macrotermitinae). Insectes Sociaux 1983, 30, 204–209. [Google Scholar] [CrossRef]
- Aanen, D.K.; Eggleton, P.; Rouland-Lefevre, C.; Guldberg-Frøslev, T.; Rosendahl, S.; Boomsma, J.J. The evolution of fungus-growing termites and their mutualistic fungal symbionts. Proc. Natl. Acad. Sci. USA 2002, 99, 14887–14892. [Google Scholar] [CrossRef] [PubMed]
- Nobre, T.; Kone, N.A.; Konate, S.; Linsenmair, K.E.; Aanen, D.K. Dating the fungus-growing termites’ mutualism shows a mixture between ancient codiversification and recent symbiont dispersal across divergent hosts. Mol. Ecol. 2011, 20, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Brandl, R.; Hyodo, F.; von Korff-Schmising, M.; Maekawa, K.; Miura, T.; Takematsu, Y.; Matsumoto, T.; Abe, T.; Bagine, R.; Kaib, M. Divergence times in the termite genus Macrotermes (Isoptera: Termitidae). Mol. Phylogenetics Evol. 2007, 45, 239–250. [Google Scholar] [CrossRef]
- Aanen, D.K.; Eggleton, P. Fungus-growing termites originated in African rain forest. Curr. Biol. 2005, 15, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Maekawa, K.; Kitade, O.; Abe, T.; Matsumoto, T. Phylogenetic relationships among subfamilies in higher termites (Isoptera Termitidae) based on mitochondrial COII gene sequences. Ann. Entomol. Soc. Am. 1998, 91, 515–523. [Google Scholar] [CrossRef]
- Donovan, S.; Jones, D.; Sands, W.; Eggleton, P. Morphological phylogenetics of termites (Isoptera). Biol. J. Linn. Soc. 2000, 70, 467–513. [Google Scholar] [CrossRef]
- Kambhampati, S.; Eggleton, P. Taxonomy and phylogeny of termites. In Termites: Evolution, Sociality, Symbioses, Ecology; Springer: Berlin/Heidelberg, Germany, 2000; pp. 1–23. [Google Scholar]
- AlShamakhi, H.S.; Al-Sadi, A.M.; Cook, L.G. Phylogenetic Relationships of the Mutualistic Fungi Associated with Macrotermes subhyalinus in Oman. Mycobiology 2023, 51, 281–287. [Google Scholar] [CrossRef]
- Eggleton, P. Global patterns of termite diversity. In Termites: Evolution, Sociality, Symbioses, Ecology; Springer: Berlin/Heidelberg, Germany, 2000; pp. 25–51. [Google Scholar]
- Darlington, J.P.E.C. Nutrition and evolution in fungus-growing termites. In Nourishment and Evolution in Insect Societies; Hunt, J.H., Nalepa, C.A., Eds.; Westview Press, Inc.: Boulder, CO, USA, 1994; pp. 105–130. [Google Scholar]
- Wang, M.-M.; Song, N.; Guo, S.-B.; Yin, X.-M. A comprehensive sampling of mitogenomes shows the utility to infer phylogeny of termites (Blattodea: Termitoidae). J. Insect Sci. 2024, 24, 7. [Google Scholar] [CrossRef]
- Qiu, B.; Elsner, D.; Korb, J. High-quality long-read genome assemblies reveal evolutionary patterns of transposable elements and DNA methylation in termites. bioRxiv 2023, preprint. [Google Scholar] [CrossRef]
- Ruelle, J.E. A Revision of the Termites of the Genus Macrotermes from the Ethiopian Region (Isoptera: Termitidae); British Museum (Natural History): London, UK, 1970; pp. 366–444. [Google Scholar]
- Bagine, R.; Darlington, J.; Kat, P.; Ritchie, J. Nest structure, population structure and genetic differentiation of some morphologically similar species of Macrotermes in Kenya. Sociobiology 1989, 15, 125–132. [Google Scholar]
- Egan, B.; Nethavhani, Z.; van Asch, B. Overview of the genetic diversity of African Macrotermes (Termitidae: Macrotermitinae) and implications for taxonomy, ecology and food science. Insects 2021, 12, 518. [Google Scholar] [CrossRef]
- van der Werff, P.A. Two mound types of Macrotermes near Kajiado (Kenya): Intraspecific variation or interspecific divergence? In Biosystematics of Social Insects; Howse, P.E., Clement, J.L., Eds.; Academic Press: New York, NY, USA, 1981; Volume 19, pp. 231–247. [Google Scholar]
- Marten, A.; Kaib, M.; Brandl, R. Cuticular hydrocarbon phenotypes do not indicate cryptic species in fungus-growing termites (Isoptera: Macrotermitinae). J. Chem. Ecol. 2009, 35, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Rambur, P. Histoire Naturelle des Insectes. Névroptères; Roret: Paris, France, 1842. [Google Scholar]
- Haviland, G.D. Observations on termites; with descriptions of new species. J. Linn. Soc. Lond. Zool. 1898, 26, 358–442. [Google Scholar] [CrossRef]
- Silvestri, F. Contribuzione alla conoscenza dei Termitidae e Termitofili dell’Africa occidentale. 1. Termitidi. Boll. Del Lab. Zool. Gen. E Agrar. 1914, 1–146. [Google Scholar]
- Darlington, J.P.E.C. Two types of mound built by the termite Macrotermes subhyalinus in Kenya. Int. J. Trop. Insect Sci. 1984, 5, 481–492. [Google Scholar] [CrossRef]
- Sjostedt, Y. Isoptera in: W. Michaelsen. Beitr. Land-u. Siiswasserfauna Dtsch-SiidwAfr 1914, 1, 73–92. [Google Scholar]
- Inward, D.J.; Vogler, A.P.; Eggleton, P. A comprehensive phylogenetic analysis of termites (Isoptera) illuminates key aspects of their evolutionary biology. Mol. Phylogenet. Evol. 2007, 44, 953–967. [Google Scholar] [CrossRef]
- Dowton, M.; Austin, A. Phylogenetic relationships among the microgastroid wasps (Hymenoptera: Braconidae): Combined analysis of 16S and 28S rDNA genes and morphological data. Mol. Phylogenetics Evol. 1998, 10, 354–366. [Google Scholar] [CrossRef]
- Whiting, M.F.; Carpenter, J.C.; Wheeler, Q.D.; Wheeler, W.C. The Strepsiptera problem: Phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst. Biol. 1997, 46, 1–68. [Google Scholar] [CrossRef]
- Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Lin, Y.P.; Edwards, R.D.; Kondo, T.; Semple, T.L.; Cook, L.G. Species delimitation in asexual insects of economic importance: The case of black scale (Parasaissetia nigra), a cosmopolitan parthenogenetic pest scale insect. PLoS ONE 2017, 12, e0175889. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (* and other Methods), 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Bull, J.J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Maddison, D.R. The discovery and importance of multiple islands of most-parsimonious trees. Syst. Biol. 1991, 40, 315–328. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T.-a. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Rambaut, A.; Suchard, M.; Xie, D.; Drummond, A. Tracer v1. 6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 12 August 2024).
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Rozas, J.; Rozas, R. DnaSP, DNA sequence polymorphism: An interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. CABIOS 1995, 11, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Dorman, K.S.; Fang, F.; Suchard, M.A. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics 2005, 21, 3034–3042. [Google Scholar] [CrossRef]
- Suchard, M.A.; Weiss, R.E.; Dorman, K.S.; Sinsheimer, J.S. Inferring Spatial Phylogenetic Variation Along Nucleotide Sequences: A Multiple Changepoint Model. J. Am. Stat. Assoc. 2003, 98, 427–437. [Google Scholar] [CrossRef]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol. 2006, 15, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.A.; McLenachan, P.; Phillips, M.; Slack, K.E.; Cooper, A.; Penny, D. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol. Biol. Evol. 2004, 21, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenetics Evol. 2003, 28, 171–185. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Jermiin, L.S.; Ho, S.Y.; Ababneh, F.; Robinson, J.; Larkum, A.W. The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst. Biol. 2004, 53, 638–643. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Korb, J.; Kasseney, B.D.; Cakpo, Y.T.; Casalla Daza, R.H.; Gbenyedji, J.N.K.; Ilboudo, M.E.; Josens, G.; Koné, N.g.A.; Meusemann, K.; Ndiaye, A.B. Termite taxonomy, challenges and prospects: West Africa, a case example. Insects 2019, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Montelongo, T.; Gómez-Zurita, J. Multilocus molecular systematics and evolution in time and space of Calligrapha (Coleoptera: Chrysomelidae, Chrysomelinae). Zool. Scr. 2014, 43, 605–628. [Google Scholar] [CrossRef]
- Quinzin, M.C.; Mardulyn, P. Multi-locus DNA sequence variation in a complex of four leaf beetle species with parapatric distributions: Mitochondrial and nuclear introgressions reveal recent hybridization. Mol. Phylogenetics Evol. 2014, 78, 14–24. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Soldier Head Capsule | Distinctive Features | Mound Structure | Swarming Behavior | Known Distribution | ||
|---|---|---|---|---|---|---|
| M. falciger | Measurements: 142 specimens from 64 localities, range is in millimeters Length (5.20–7.43) Width (4.00–6.14) Length of left mandible (2.85–4.01) Max. length of pronotum (1.70–261) Max. width of pronotum (3.30–5.18) | Colors: Dark chestnut-brown | Major soldiers of M. falciger are usually darker and are characterized by huge thoracic nota, a wide head, thick antennae, and conspicuous pilosity of the gula The minor soldiers have longer tibiae and more incurved mandibles than in M. subhyalinus Imagos of M. falciger are usually larger than those of M. subhyalinus and have proportionately smaller eyes and longer tibiae, and the depressed area around the fontanelle also differs in shape | Low hummocks, but can vary under different circumstances | Just after dark | Predominantly a woodland species, which is widely distributed in eastern and southern parts of Africa |
| M. subhyalinus | Measurements: 456 specimens from 163 localities, range is in millimeters Length (4.13–6.21) Width (3.26–4.89) Length of left mandible (2.52–3.14) Max. length of pronotum (1.43–1.90) Max. width of pronotum (2.55–3.74) | Red–yellow to reddish brown | Erects tall and spired, but can vary under different circumstances | Around midnight | Widely distributed through West, Central, and East Africa | |
| Mean | A | C | G | T | Sites | Variable Sites | Among Taxa | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Chi-Square | df | p > 0.05 | ||||||||
| Macrotermitinae | 1st-codon position | 0.32415 | 0.23234 | 0.27605 | 0.16746 | 179.00 | 42 | 24.909676 | 264 | 1.00000000 |
| 2nd-codon position | 0.13723 | 0.27883 | 0.17032 | 0.41362 | 178.99 | 15 | 3.245748 | 264 | 1.00000000 | |
| 3rd-codon position | 0.48886 | 0.28916 | 0.07286 | 0.14912 | 178.99 | 168 | 249.222679 | 264 | 0.81496609 | |
| All codon positions | 0.31661 | 0.26682 | 0.17320 | 0.24337 | 536.97 | 225 | 78.074355 | 264 | 1.00000000 | |
| Macrotermitinae and outgroup | 1st-codon position | 0.32537 | 0.22856 | 0.27486 | 0.17121 | 179.00 | 49 | 53.197546 | 354 | 1.00000000 |
| 2nd-codon position | 0.13667 | 0.27773 | 0.17071 | 0.41488 | 178.99 | 17 | 4.698318 | 354 | 1.00000000 | |
| 3rd-codon position | 0.48925 | 0.27340 | 0.06934 | 0.16801 | 178.99 | 169 | 518.419339 | 354 | 0.00000003 | |
| All codon positions | 0.31705 | 0.25988 | 0.17167 | 0.25140 | 536.98 | 235 | 194.923233 | 354 | 1.00000000 | |
| Outgroup only | 1st-codon position | 0.32880 | 0.21623 | 0.27132 | 0.18365 | 179.00 | 30 | 17.396548 | 87 | 1.00000000 |
| 2nd-codon position | 0.13501 | 0.27412 | 0.17188 | 0.41899 | 179.00 | 3 | 0.638419 | 87 | 1.00000000 | |
| 3rd-codon position | 0.49300 | 0.22388 | 0.05703 | 0.22609 | 179.00 | 154 | 60.210541 | 87 | 0.98730157 | |
| All codon positions | 0.31880 | 0.23802 | 0.16683 | 0.27634 | 537.00 | 187 | 24.159910 | 87 | 1.00000000 | |
| RY coding: Macrotermitinae and outgroup | 1st-codon position | 0.32537 | 0.22856 | 0.27486 | 0.17121 | 178.97 | 49 | 53.196584 | 354 | 1.00000000 |
| 2nd-codon position | 0.13667 | 0.27773 | 0.17071 | 0.41488 | 178.98 | 17 | 4.698318 | 354 | 1.00000000 | |
| 3rd-codon position | 0.27928 | 0.22072 | 0.27982 | 0.22072 | 178.98 | 104 | 38.248258 | 354 | 1.00000000 | |
| All codon positions | 0.24879 | 0.23697 | 0.23992 | 0.27432 | 536.94 | 170 | 47.366512 | 354 | 1.00000000 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlShamakhi, H.S.; Al-Sadi, A.M.; Cook, L.G. Evolutionary Relationships of Omani Macrotermes subhyalinus, Macrotermitinae. Insects 2024, 15, 648. https://doi.org/10.3390/insects15090648
AlShamakhi HS, Al-Sadi AM, Cook LG. Evolutionary Relationships of Omani Macrotermes subhyalinus, Macrotermitinae. Insects. 2024; 15(9):648. https://doi.org/10.3390/insects15090648
Chicago/Turabian StyleAlShamakhi, Hilal S., Abdullah M. Al-Sadi, and Lyn G. Cook. 2024. "Evolutionary Relationships of Omani Macrotermes subhyalinus, Macrotermitinae" Insects 15, no. 9: 648. https://doi.org/10.3390/insects15090648