
Host Species Affects Gut Microbial Community and Offspring Developmental Performances in the Pupal Parasitoid Chouioia cunea Yang (Hymenoptera: Eulophidae)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Rearing and Development Study
2.2. Sample Collection and DNA Extraction
2.3. 16S rRNA Gene Amplification and Sequencing
2.4. OTU Identification and Diversity Analysis
2.5. Statistical Analysis
3. Results
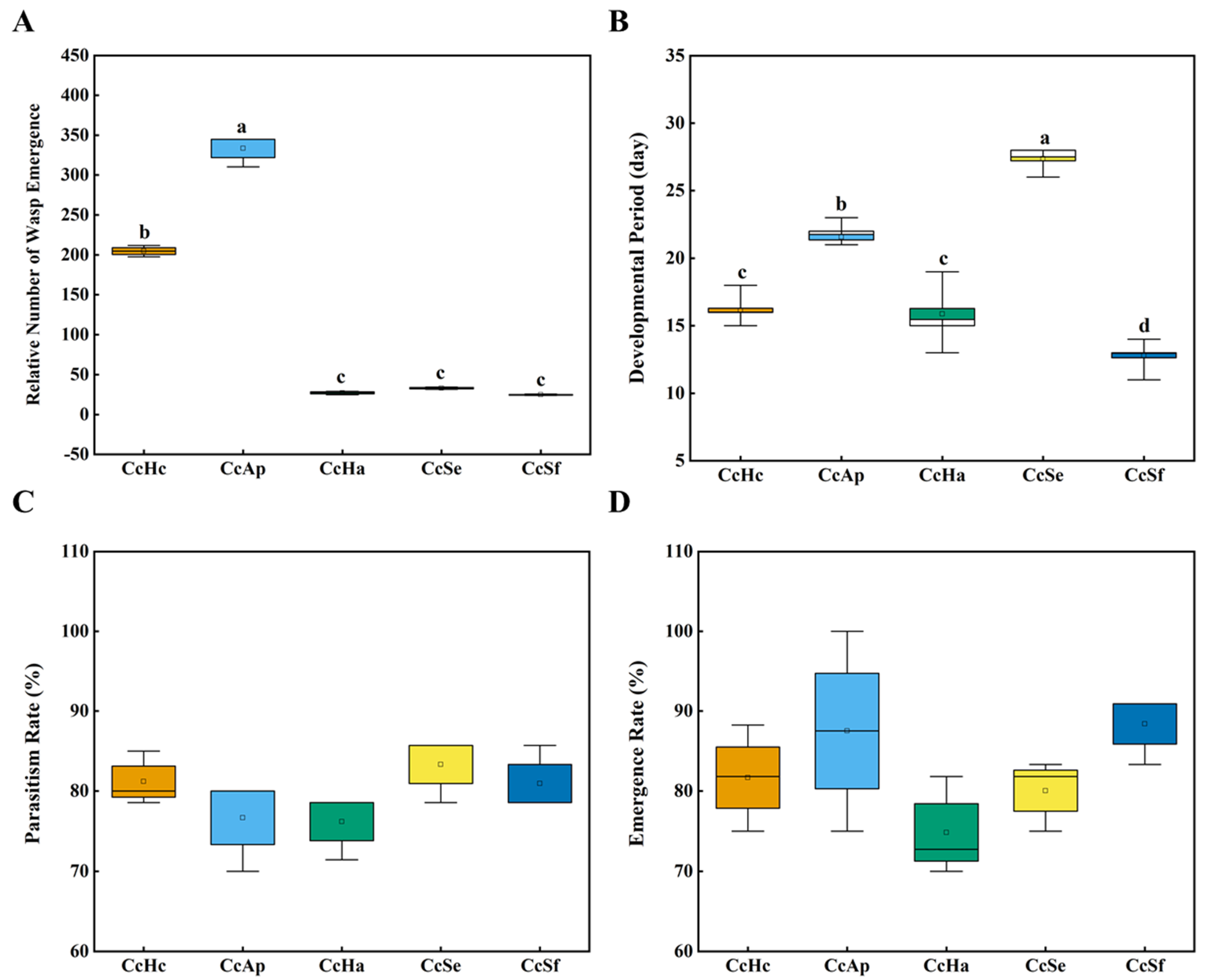
3.1. The Parasitic Capability and Development of C. cunea among Different Hosts
3.2. Sequencing Data of 16S rRNA
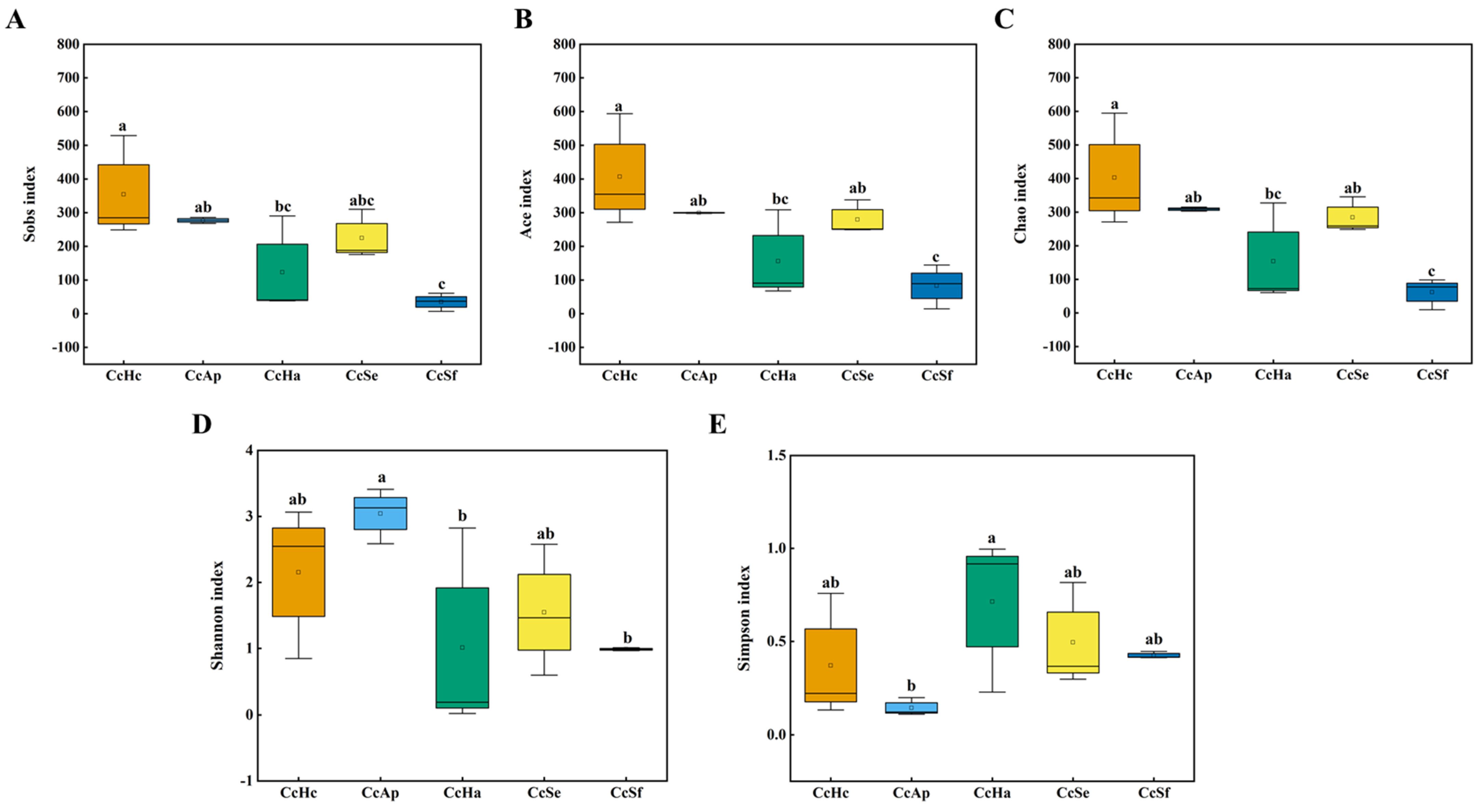
3.3. Gut Microbial Diversity of C. cunea among Different Hosts
3.4. Gut Microbial Composition of C. cunea among Different Hosts
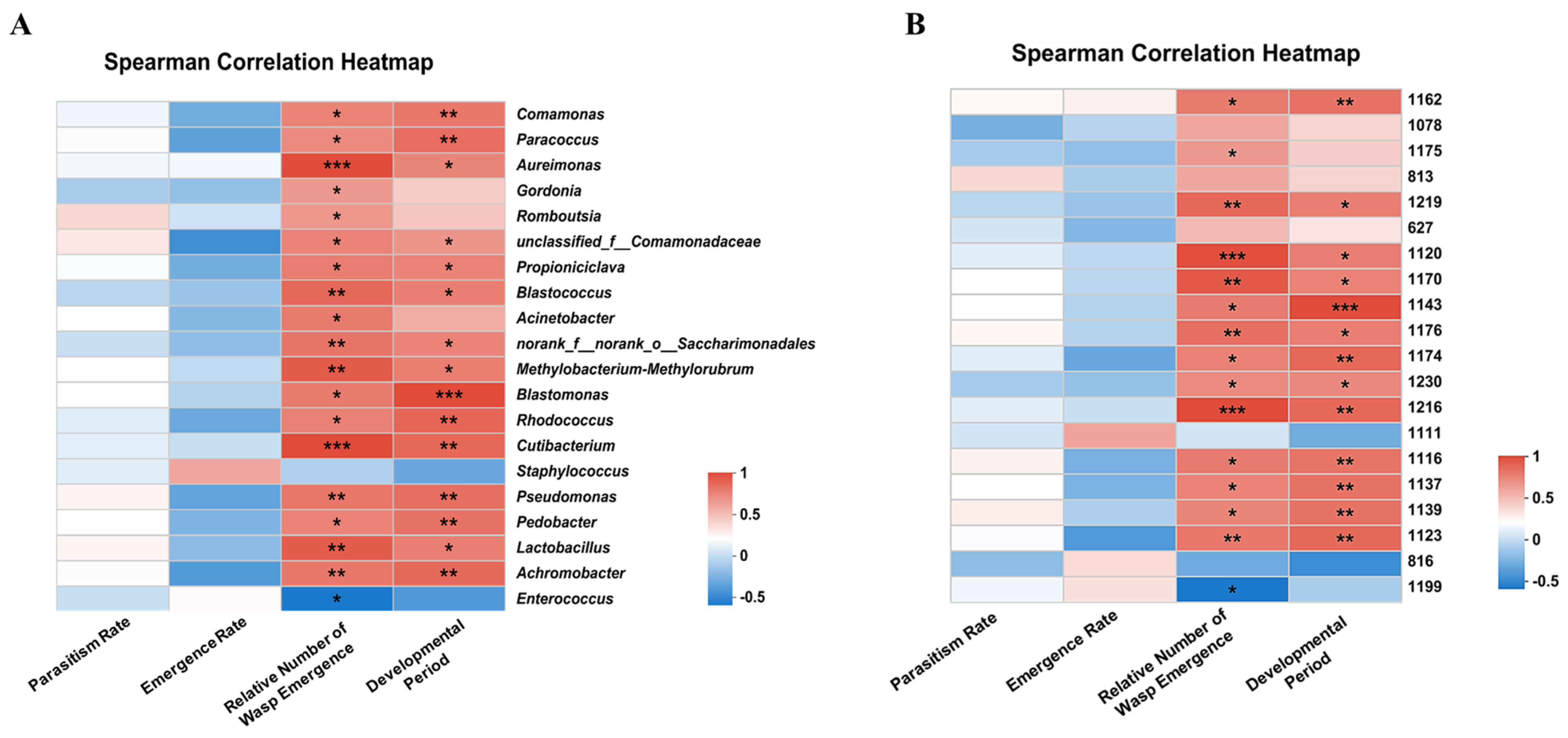
3.5. Correlation Analysis of Intestinal Microbial Abundance and Development of C. cunea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Nair, S. Dynamics of insect–microbiome interaction influence host and microbial symbiont. Front. Microbiol. 2020, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Ravigné, V.; Becker, N.; Massol, F.; Guichoux, E.; Boury, C.; Mahé, F.; Facon, B. Fruit fly phylogeny imprints bacterial gut microbiota. Evol. Appl. 2022, 15, 1621–1638. [Google Scholar] [CrossRef] [PubMed]
- Grieco, M.B.; Lopes, F.A.C.; Oliveira, L.S.; Tschoeke, D.A.; Popov, C.C.; Thompson, C.C.; Goncalves, L.C.; Constantino, R.; Martins, O.B.; Kruger, R.H.; et al. Metagenomic Analysis of the whole gut microbiota in brazilian termitidae termites Cornitermes cumulans, Cyrilliotermes strictinasus, Syntermes dirus, Nasutitermes jaraguae, Nasutitermes aquilinus, Grigiotermes bequaerti, and Orthognathotermes mirim. Curr. Microbiol. 2019, 76, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2020, 19, 241–255. [Google Scholar] [CrossRef]
- Li, D.-D.; Li, J.-Y.; Hu, Z.-Q.; Liu, T.-X.; Zhang, S.-Z. Fall armyworm gut bacterial diversity associated with different developmental stages, environmental habitats, and diets. Insects 2022, 13, 762. [Google Scholar] [CrossRef]
- Gong, Q.; Cao, L.-J.; Sun, L.-N.; Chen, J.-C.; Gong, Y.-J.; Pu, D.-Q.; Huang, Q.; Hoffmann, A.A.; Wei, S.-J. Similar gut bacterial microbiota in two fruit-feeding moth pests collected from different host species and locations. Insects 2020, 11, 840. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, X.; Liu, X.; Dong, Y.; Yan, Z.; Lv, D.; Wang, P.; Li, Y. Comparison of gut bacterial communities of Grapholita molesta (Lepidoptera: Tortricidae) reared on different host plants. Int. J. Mol. Sci. 2021, 22, 6843. [Google Scholar] [CrossRef]
- Yang, F.-Y.; Saqib, H.S.A.; Chen, J.-H.; Ruan, Q.-Q.; Vasseur, L.; He, W.-Y.; You, M.-S. Differential profiles of gut microbiota and metabolites associated with host shift of Plutella xylostella. Int. J. Mol. Sci. 2020, 21, 6283. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Zhang, X.; Yun, Y. Diversity of bacteria associated with guts and gonads in three spider species and potential transmission pathways of microbes within the same spider host. Insects 2023, 14, 792. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, P.; Qin, W.; Li, H.; Zheng, J.; Meng, L.; Li, B. Gut microbiota variation across generations regarding the diet and life stage in Harmonia axyridis (Coleoptera: Coccinellidae). Insect Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Brinker, P.; Chen, F.; Chehida, Y.B.; Beukeboom, L.W.; Fontaine, M.C.; Salles, J.F. Microbiome composition is shaped by geography and population structure in the parasitic wasp Asobara japonica, but not in the presence of the endosymbiont Wolbachia. Mol. Ecol. 2022, 32, 6644–6658. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Xu, H.; Gao, S.; Gao, Z.; Wang, N. Effects of different hosts on bacterial communities of parasitic wasp Nasonia vitripennis. Front. Microbiol. 2020, 11, 1435. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Wei, J.; Wang, X. Studies on alternate hosts of the parasiotid Chouioia cunea (Hymenoptera: Eulophidae). Sci. Silvae Sin. 2004, 40, 106–116. [Google Scholar]
- Zheng, Y.; Qi, J.; Sun, S.; Yang, C. Advance in research of Chouioia cunea Yang (Hymenoptera: Eulophidade) and its biocontrol application in China. Chin. J. Biol. Control 2012, 28, 275–281. [Google Scholar] [CrossRef]
- Pan, L.; Gao, W.; Liu, X.; Qin, D.; Zhang, T.; Ren, R.; Zhang, W.; Sun, M.; Gao, C.; Bai, P.; et al. Parasitoids as taxonomists: How does the parasitoid Chouioia cunea distinguish between a host and a non-host? Pest Manag. Sci. 2023, 79, 4547–4556. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. vegan: Community Ecology Package, Version 2.5-3. 2001. Available online: https://CRAN.R-project.org/package=vegan (accessed on 19 April 2023).
- Li, M.; Yang, Y.; Yao, Y.; Xiang, W.; Han, J.; Wang, Y.; Bai, P.; Wang, J.; Zhu, G.; Man, L.; et al. Isolation and identification of attractants from the pupae of three lepidopteran species for the parasitoid Chouioia cunea Yang. Pest Manag. Sci. 2020, 76, 1920–1928. [Google Scholar] [CrossRef]
- Runge, S.; Rosshart, S.P. The mammalian metaorganism: A holistic view on how microbes of all kingdoms and niches shape local and systemic immunity. Front. Immunol. 2021, 12, 702378. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Moran, N.A. The role of the gut microbiome in health and disease of adult honey bee workers. Curr. Opin. Insect Sci. 2018, 26, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Rahman, M.-M.; Han, C.; Shin, J.; Sa, K.J.; Kim, J. Spodoptera frugiperda (Lepidoptera: Noctuidae) life table comparisons and gut microbiome analysis reared on corn varieties. Insects 2023, 14, 358. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Du, K.; Sun, C.; Vimalanathan, A.; Liang, X.; Li, Y.; Wang, B.; Lu, X.; Li, L.; Shao, Y. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 2018, 12, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef]
- Kudo, R.; Masuya, H.; Endoh, R.; Kikuchi, T.; Ikeda, H. Gut bacterial and fungal communities in ground-dwelling beetles are associated with host food habit and habitat. ISME J. 2019, 13, 676–685. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, Z.; Yu, J.; Li, Z.; Liu, X.; Xu, H. Comparison of gut bacterial communities and their associations with host diets in four fruit borers. Pest Manag. Sci. 2019, 76, 1353–1362. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, J.; Tan, H.; Fu, Z.; Wang, X. Characterization of the gut microbiome in the beet armyworm Spodoptera exigua in response to the short-term thermal stress. J. Asia-Pac. Entomol. 2022, 25, 101863. [Google Scholar] [CrossRef]
- Gao, H.; Jiang, S.; Wang, Y.; Hu, M.; Xue, Y.; Cao, B.; Dou, H.; Li, R.; Yi, X.; Jiang, L.; et al. Comparison of gut bacterial communities of Hyphantria cunea Drury (Lepidoptera, Arctiidae), based on 16S rRNA full-length sequencing. Biodivers. Data J. 2023, 11, e98143. [Google Scholar] [CrossRef]
- Tian, Z.; Guo, X.; Michaud, J.P.; Zha, M.; Zhu, L.; Liu, X.; Liu, X. The gut microbiome of Helicoverpa armigera enhances immune response to baculovirus infection via suppression of Duox-mediated reactive oxygen species. Pest Manag. Sci. 2023, 79, 3611–3621. [Google Scholar] [CrossRef]
- Wang, X.; Sun, S.; Yang, X.; Cheng, J.; Wei, H.; Li, Z.; Michaud, J.P.; Liu, X. Variability of gut microbiota across the life cycle of Grapholita molesta (Lepidoptera: Tortricidae). Front. Microbiol. 2020, 11, 1366. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Sun, B.; Gurr, G.M.; Vasseur, L.; Xue, M.; You, M. Gut microbiota mediate insecticide resistance in the diamondback moth, Plutella xylostella (L.). Front. Microbiol. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Zhang, Y.; Sun, Z.; Li, Y.; Ding, J.; Zhang, G.; Du, E.; Zi, X.; Tian, C.; et al. Role of Enterococcus mundtii in gut of the tomato leaf miner (Tuta absoluta) to detoxification of Chlorantraniliprole. Pestic. Biochem. Physiol. 2024, 204, 106060. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hao, D.; Chen, C.; Sun, Y.; Yu, X.; Bextine, B. Effects of midgut bacteria in Hyphantria cunea(Lepidoptera: Erebidae) on nuclear polyhedrosis virus and Bacillus thuringiensis (Bacillales: Bacillaceae). J. Insect Sci. 2023, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ni, B.; Wu, Y.; Yang, Y.; Mu, D.; Wu, K.; Zhang, A.; Du, Y.; Li, Q. The cultivable gut bacteria Enterococcus mundtii promotes early-instar larval growth of Conogethes punctiferalis via enhancing digestive enzyme activity. Pest Manag. Sci. 2024. [Google Scholar] [CrossRef]
- Bing, X.L.; Winkler, J.; Gerlach, J.; Loeb, G.; Buchon, N. Identification of natural pathogens from wild Drosophila suzukii. Pest Manag. Sci. 2021, 77, 1594–1606. [Google Scholar] [CrossRef]
- Schmidt, K.; Engel, P. Mechanisms underlying gut microbiota–host interactions in insects. J. Exp. Biol. 2021, 224, jeb207696. [Google Scholar] [CrossRef]
- Chopra, I.; Schofield, C.; Everett, M.; O’Neill, A.; Miller, K.; Wilcox, M.; Frère, J.-M.; Dawson, M.; Czaplewski, L.; Urleb, U.; et al. Treatment of health-care-associated infections caused by Gram-negative bacteria: A consensus statement. Lancet Infect. Dis. 2008, 8, 133–139. [Google Scholar] [CrossRef]
- Srinatha, H.S.; Jalali, S.K.; Sriram, S.; Chakravarthy, A.K. Isolation of microbes associated with field-collected populations of the egg parasitoid, Trichogramma chiloniscapable of enhancing biotic fitness. Biocontrol Sci. Technol. 2015, 25, 789–802. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, L.; Liao, J.; Hu, Y.; Ren, R.; Chen, W.; Liang, Z.; Lu, F.; Sun, M.; Song, Z.; Li, X.; et al. Host Species Affects Gut Microbial Community and Offspring Developmental Performances in the Pupal Parasitoid Chouioia cunea Yang (Hymenoptera: Eulophidae). Insects 2024, 15, 722. https://doi.org/10.3390/insects15090722
Pan L, Liao J, Hu Y, Ren R, Chen W, Liang Z, Lu F, Sun M, Song Z, Li X, et al. Host Species Affects Gut Microbial Community and Offspring Developmental Performances in the Pupal Parasitoid Chouioia cunea Yang (Hymenoptera: Eulophidae). Insects. 2024; 15(9):722. https://doi.org/10.3390/insects15090722
Chicago/Turabian StylePan, Lina, Jiamin Liao, Yiping Hu, Rui Ren, Wei Chen, Zixin Liang, Fan Lu, Meidi Sun, Zhiqin Song, Xiaoyu Li, and et al. 2024. "Host Species Affects Gut Microbial Community and Offspring Developmental Performances in the Pupal Parasitoid Chouioia cunea Yang (Hymenoptera: Eulophidae)" Insects 15, no. 9: 722. https://doi.org/10.3390/insects15090722
APA StylePan, L., Liao, J., Hu, Y., Ren, R., Chen, W., Liang, Z., Lu, F., Sun, M., Song, Z., Li, X., Zhang, W., Gao, W., Yan, C., & Li, M. (2024). Host Species Affects Gut Microbial Community and Offspring Developmental Performances in the Pupal Parasitoid Chouioia cunea Yang (Hymenoptera: Eulophidae). Insects, 15(9), 722. https://doi.org/10.3390/insects15090722