

Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins


Abstract
1. Introduction
2. Results
2.1. Difference Between EFI and Correlation Coefficient
2.2. Methylation Sites Are Linked to Disorders
2.3. Environmental Factors Alter DNA Methylation Levels in Methylation Markers
2.4. DNA Methylation on CpG Islands
3. Discussion
4. Materials and Methods
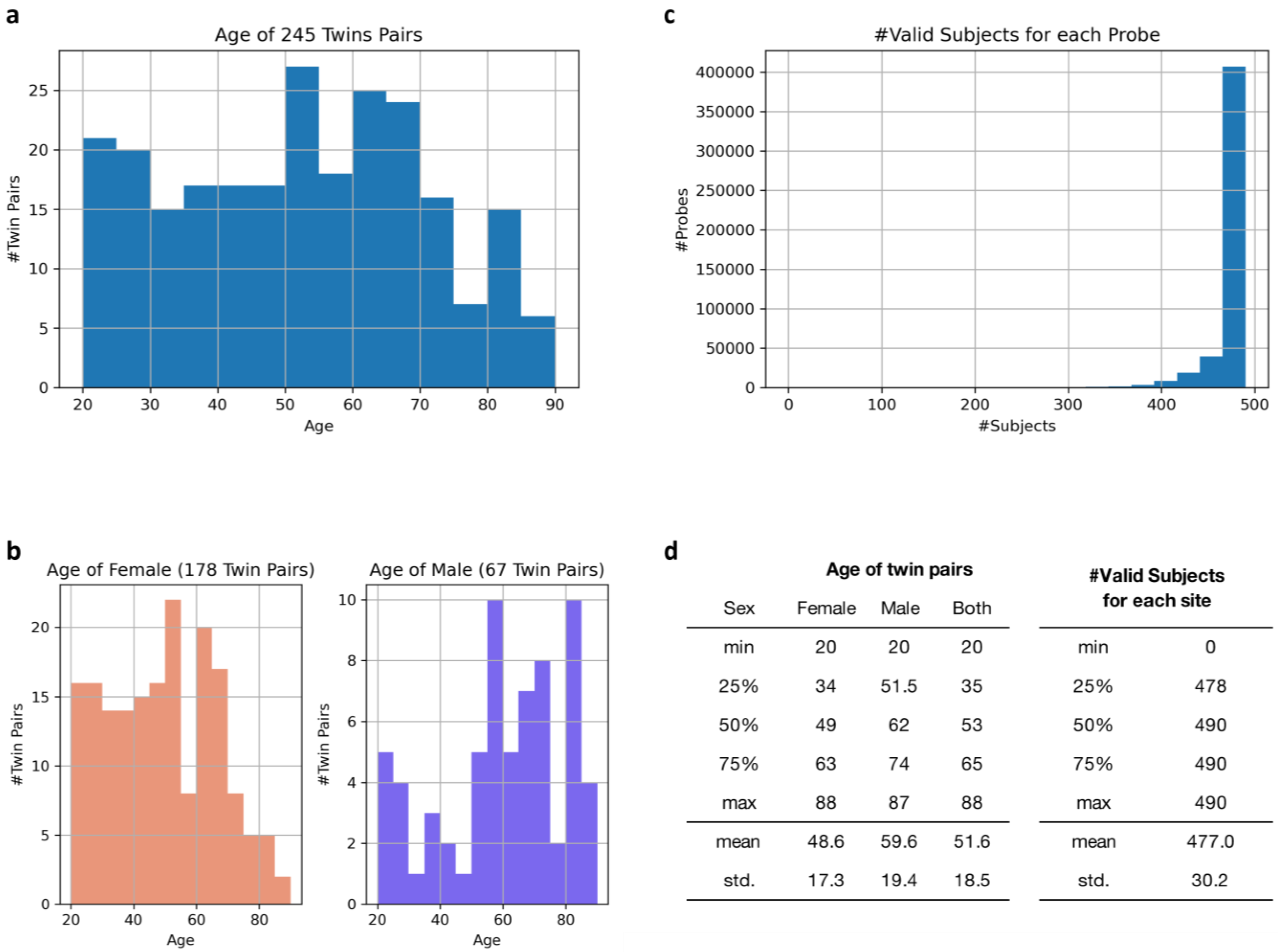
4.1. Subjects and Ethics Statement
4.2. Methylation Sites
4.3. Notation
4.4. Environmental Factor Index (EFI)
4.5. Statistical Analysis of EFI
4.6. Statistical Analysis of Disease Association
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Dunn, D.; Smith, J. The occurrence of 6-methylaminopurine in deoxyribonucleic acids. Biochem. J. 1958, 68, 627. [Google Scholar] [CrossRef]
- Vanyushin, B.; Tkacheva, S.; Belozersky, A. Rare bases in animal DNA. Nature 1970, 225, 948–949. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Carreira, L.H.; Ljungdahl, L.G.; Kuo, K.C.; Gehrke, C.W. DNA methylation in thermophilic bacteria: N 4-methylcytosine, 5-methylcytosine, and N 5 methyladenine. Nucleic Acids Res. 1985, 13, 1399–1412. [Google Scholar] [CrossRef]
- Ratel, D.; Ravanat, J.L.; Berger, F.; Wion, D. N6-methyladenine: The other methylated base of DNA. Bioessays 2006, 28, 309–315. [Google Scholar] [CrossRef]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M. DNA methylation on N 6-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Estève, P.-O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef]
- Bender, C.M.; Gonzalgo, M.L.; Gonzales, F.A.; Nguyen, C.T.; Robertson, K.D.; Jones, P.A. Roles of cell division and gene transcription in the methylation of CpG islands. Mol. Cell. Biol. 1999, 19, 6690–6698. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef]
- MüLLER, H.M.; Fiegl, H.; Widschwendter, A.; Widschwendter, M. Prognostic DNA methylation marker in serum of cancer patients. Ann. N. Y. Acad. Sci. 2004, 1022, 44–49. [Google Scholar] [CrossRef]
- Sigin, V.O.; Kalinkin, A.I.; Kuznetsova, E.B.; Simonova, O.A.; Chesnokova, G.G.; Litviakov, N.V.; Slonimskaya, E.M.; Tsyganov, M.M.; Ibragimova, M.K.; Volodin, I.V. DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer. Sci. Rep. 2020, 10, 9239. [Google Scholar] [CrossRef]
- Xu, W.; Xu, M.; Wang, L.; Zhou, W.; Xiang, R.; Shi, Y.; Zhang, Y.; Piao, Y. Integrative analysis of DNA methylation and gene expression identified cervical cancer-specific diagnostic biomarkers. Signal Transduct. Target. Ther. 2019, 4, 55. [Google Scholar] [CrossRef]
- Lofton-Day, C.; Model, F.; DeVos, T.; Tetzner, R.; Distler, J.; Schuster, M.; Song, X.; Lesche, R.; Liebenberg, V.; Ebert, M. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem. 2008, 54, 414–423. [Google Scholar] [CrossRef]
- Tänzer, M.; Balluff, B.; Distler, J.; Hale, K.; Leodolter, A.; Röcken, C.; Molnar, B.; Schmid, R.; Lofton-Day, C.; Schuster, T. Performance of epigenetic markers SEPT9 and ALX4 in plasma for detection of colorectal precancerous lesions. PLoS ONE 2010, 5, e9061. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, J.; Sidransky, D. DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 2010, 29, 181–206. [Google Scholar] [CrossRef]
- Diaz-Lagares, A.; Mendez-Gonzalez, J.; Hervas, D.; Saigi, M.; Pajares, M.J.; Garcia, D.; Crujerias, A.B.; Pio, R.; Montuenga, L.M.; Zulueta, J. A novel epigenetic signature for early diagnosis in lung cancer. Clin. Cancer Res. 2016, 22, 3361–3371. [Google Scholar] [CrossRef]
- Yan, P.; Yang, X.; Wang, J.; Wang, S.; Ren, H. A novel CpG island methylation panel predicts survival in lung adenocarcinomas. Oncol. Lett. 2019, 18, 1011–1022. [Google Scholar] [CrossRef]
- Li, M.; Zhang, C.; Zhou, L.; Li, S.; Cao, Y.J.; Wang, L.; Xiang, R.; Shi, Y.; Piao, Y. Identification and validation of novel DNA methylation markers for early diagnosis of lung adenocarcinoma. Mol. Oncol. 2020, 14, 2744–2758. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B. Identification of type 1 diabetes–associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef]
- Davegårdh, C.; García-Calzón, S.; Bacos, K.; Ling, C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol. Metab. 2018, 14, 12–25. [Google Scholar] [CrossRef]
- Ahmed, S.A.H.; Ansari, S.A.; Mensah-Brown, E.P.; Emerald, B.S. The role of DNA methylation in the pathogenesis of type 2 diabetes mellitus. Clin. Epigenetics 2020, 12, 104. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Müller, G.A.; Kalbacher, H.; Salant, D.J.; Müller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.-A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.-L.; Rantapää-Dahlqvist, S.; Bengtsson, A.A.; Jönsen, A.; Padyukov, L. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Sjöwall, C.; Syvänen, A.-C.; Rönnblom, L.; Sandling, J.K.; Nordmark, G. Shared and Unique Patterns of DNA Methylation in Systemic Lupus Erythematosus and Primary Sjögren’s Syndrome. Front. Immunol. 2019, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Edris, A.; den Dekker, H.T.; Melén, E.; Lahousse, L. Epigenome-wide association studies in asthma: A systematic review. Clin. Exp. Allergy 2019, 49, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Alag, A. Machine learning approach yields epigenetic biomarkers of food allergy: A novel 13-gene signature to diagnose clinical reactivity. PLoS ONE 2019, 14, e0218253. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Kim, M.K.; Sasaki, S.; Otani, T.; Tsugane, S.; Japan Public Health Center-based Prospective Study Group. Dietary patterns and subsequent colorectal cancer risk by subsite: A prospective cohort study. Int. J. Cancer 2005, 115, 790–798. [Google Scholar] [CrossRef]
- Wang, K.; Gaitsch, H.; Poon, H.; Cox, N.J.; Rzhetsky, A. Classification of common human diseases derived from shared genetic and environmental determinants. Nat. Genet. 2017, 49, 1319. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Keil, K.P.; Lein, P.J. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenetics 2016, 2, dvv012. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.-H.; Maekawa, T.; Ishii, S. Inheritance of Stress-Induced Epigenetic Changes Mediated by the ATF-2 Family of Transcription Factors. In Stress-Induced Mutagenesis; Springer: Berlin/Heidelberg, Germany, 2013; pp. 103–118. [Google Scholar]
- Radford, E.J.; Ito, M.; Shi, H.; Corish, J.A.; Yamazawa, K.; Isganaitis, E.; Seisenberger, S.; Hore, T.A.; Reik, W.; Erkek, S. In utero undernourishment perturbs the adult sperm methylome and is linked to metabolic disease transmission. Science 2014, 345, 1255903. [Google Scholar] [CrossRef]
- Bell, J.T.; Spector, T.D. A twin approach to unraveling epigenetics. Trends Genet. 2011, 27, 116–125. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Fujii, R.; Sato, S.; Tsuboi, Y.; Cardenas, A.; Suzuki, K. DNA methylation as a mediator of associations between the environment and chronic diseases: A scoping review on application of mediation analysis. Epigenetics 2022, 17, 759–785. [Google Scholar] [CrossRef]
- Burgio, E.; Piscitelli, P.; Colao, A. Environmental carcinogenesis and transgenerational transmission of carcinogenic risk: From genetics to epigenetics. Int. J. Environ. Res. Public Health 2018, 15, 1791. [Google Scholar] [CrossRef]
- Fallet, M.; Blanc, M.; Di Criscio, M.; Antczak, P.; Engwall, M.; Bosagna, C.G.; Rüegg, J.; Keiter, S.H. Present and future challenges for the investigation of transgenerational epigenetic inheritance. Environ. Int. 2023, 172, 107776. [Google Scholar] [CrossRef]
- Boomsma, D.; Busjahn, A.; Peltonen, L. Classical twin studies and beyond. Nat. Rev. Genet. 2002, 3, 872–882. [Google Scholar] [CrossRef]
- Silventoinen, K.; Rokholm, B.; Kaprio, J.; Sørensen, T.I. The genetic and environmental influences on childhood obesity: A systematic review of twin and adoption studies. Int. J. Obes. 2010, 34, 29–40. [Google Scholar] [CrossRef]
- Bell, J.T.; Saffery, R. The value of twins in epigenetic epidemiology. Int. J. Epidemiol. 2012, 41, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.; Joo, J.E.; Powell, J.E.; Ollikainen, M.; Novakovic, B.; Li, X.; Andronikos, R.; Cruickshank, M.N.; Conneely, K.N.; Smith, A.K. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res. 2012, 22, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, N.; Twik, M.; Plaschkes, I.; Nudel, R.; Iny Stein, T.; Levitt, J.; Gershoni, M.; Morrey, C.P.; Safran, M.; Lancet, D. MalaCards: An amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 2017, 45, D877–D887. [Google Scholar] [CrossRef] [PubMed]
- Behnia, F.; Parets, S.E.; Kechichian, T.; Yin, H.; Dutta, E.H.; Saade, G.R.; Smith, A.K.; Menon, R. Fetal DNA methylation of autism spectrum disorders candidate genes: Association with spontaneous preterm birth. Am. J. Obstet. Gynecol. 2015, 212, 533.e1–533.e9. [Google Scholar] [CrossRef]
- Mouat, J.S.; Li, X.; Neier, K.; Zhu, Y.; Mordaunt, C.E.; La Merrill, M.A.; Lehmler, H.J.; Jones, M.P.; Lein, P.J.; Schmidt, R.J.; et al. Networks of placental DNA methylation correlate with maternal serum PCB concentrations and child neurodevelopment. Environ. Res. 2023, 220, 115227. [Google Scholar] [CrossRef]
- Xu, X.; Yuan, X.; Ni, J.; Guo, J.; Gao, Y.; Yin, W.; Li, F.; Wei, L.; Zhang, J. MAGI2-AS3 inhibits breast cancer by downregulating DNA methylation of MAGI2. J. Cell. Physiol. 2021, 236, 1116–1130. [Google Scholar] [CrossRef]
- Qu, Y.; Gao, N.; Wu, T. Expression and clinical significance of SYNE1 and MAGI2 gene promoter methylation in gastric cancer. Medicine 2021, 100, e23788. [Google Scholar] [CrossRef]
- Chang, C.C.; Wang, H.C.; Liao, Y.P.; Chen, Y.C.; Weng, Y.C.; Yu, M.H.; Lai, H.C. The feasibility of detecting endometrial and ovarian cancer using DNA methylation biomarkers in cervical scrapings. J. Gynecol. Oncol. 2018, 29, e17. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.-P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630. [Google Scholar] [CrossRef]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Raut, J.R.; Guan, Z.; Schrotz-King, P.; Brenner, H. Fecal DNA methylation markers for detecting stages of colorectal cancer and its precursors: A systematic review. Clin. Epigenetics 2020, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33, e176. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M. Genome-wide methylation analysis of human colon cancer reveals similar hypo-and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178. [Google Scholar] [CrossRef]
- Doi, A.; Park, I.-H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S. Differential methylation of tissue-and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef]
- Cortessis, V.K.; Thomas, D.C.; Levine, A.J.; Breton, C.V.; Mack, T.M.; Siegmund, K.D.; Haile, R.W.; Laird, P.W. Environmental epigenetics: Prospects for studying epigenetic mediation of exposure–response relationships. Hum. Genet. 2012, 131, 1565–1589. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Gordon, S.D.; McRae, A.F.; Odintsova, V.V.; Mbarek, H.; Breeze, C.E.; Sugden, K.; Lundgren, S.; Castillo-Fernandez, J.E.; Hannon, E. Identical twins carry a persistent epigenetic signature of early genome programming. Nat. Commun. 2021, 12, 5618. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Caspi, A.; Williams, B.; Craig, I.W.; Houts, R.; Ambler, A.; Moffitt, T.E.; Mill, J. A longitudinal study of epigenetic variation in twins. Epigenetics 2010, 5, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Kuratomi, G.; Iwamoto, K.; Bundo, M.; Kusumi, I.; Kato, N.; Iwata, N.; Ozaki, N.; Kato, T. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Mol. Psychiatry 2008, 13, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.A.; Tang, T.; Wang, S.-C.; Ptak, C.; Oh, G.H.; Wong, A.H.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Irizarry, R.A. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc. Natl. Acad. Sci. USA 2009, 107, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Honda, C.; Watanabe, M.; Tomizawa, R.; Sakai, N.; Group, O.T.R. Update on Osaka University Twin Registry: An Overview of Multidisciplinary Research Resources and Biobank at Osaka University Center for Twin Research. Twin Res. Hum. Genet. 2019, 22, 597–601. [Google Scholar] [CrossRef]
- Watanabe, M.; Honda, C.; Iwatani, Y.; Yorifuji, S.; Iso, H.; Kamide, K.; Hatazawa, J.; Kihara, S.; Sakai, N.; Watanabe, H. Within-pair differences of DNA methylation levels between monozygotic twins are different between male and female pairs. BMC Med. Genom. 2016, 9, 55. [Google Scholar] [CrossRef]
- Dedeurwaerder, S.; Defrance, M.; Calonne, E.; Denis, H.; Sotiriou, C.; Fuks, F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011, 3, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Probe ID | Gene Symbol | EFI | q-Value | Disorder |
|---|---|---|---|---|
| cg11539424 | CLGN | 5.22 | 0.057% | |
| cg25105066 | AUTS2 | 5.14 | 0.0055% | Intellectual Developmental Disorder, Autosomal Dominant 26 |
| cg14464244 | MAGI2 | 4.76 | 0.047% | Nephrotic Syndrome, Type 15 Genetic Steroid-Resistant Nephrotic Syndrome |
| cg06445586 | 4.71 | 0.31% | ||
| cg02878907 | ZNF709 | 4.65 | 0.034% | |
| cg04883656 | OGFRL1 | 4.34 | 0.023% | |
| cg21155461 | ZNF544 | 4.04 | 0.68% | |
| cg21364278 | 4.01 | 0.37% | ||
| cg17289202 | ZNF532 | 3.95 | 0.18% | |
| cg15368722 | 3.86 | 0.084% |
| Gene Symbol | #Significant Site | Disorder |
|---|---|---|
| PTPRN2 | 39 | |
| TNXB | 34 | Ehlers–Danlos Syndrome, Classic-Like Vesicoureteral Reflux 8 |
| PRDM16 | 31 | Left Ventricular Noncompaction 8 |
| BRUNOL4 | 28 | |
| COL11A2 | 25 | Otospondylomegaepiphyseal Dysplasia, Autosomal Dominant/Recessive |
| NKX6-2 | 24 | Spastic Ataxia 8, Autosomal Recessive, with Hypomyelinating Leukodystrophy |
| PCDHGA4 | 21 | |
| THRB | 20 | Thyroid Hormone Resistance, Generalized, Autosomal Dominant Thyroid Hormone Resistance, Selective Pituitary |
| MAGI2 | 20 | Nephrotic Syndrome, Type 15 Genetic Steroid-Resistant Nephrotic Syndrome |
| TP73 | 20 | Small Cell Cancer of the Lung |
| Top 10 Sites | Top 10 Genes | Random | |
|---|---|---|---|
| #Sites | 10 | - | 100 |
| #Genes | 7 | 10 | 77 |
| #Elite genes | 2 | 7 | 19 |
| Elite gene ratio | 29% | 70% | 25% |
| p-value | 1.000 | 0.0067 | - |
| Odds ratio | 0.86 | 7.12 | - |
| #Markers | #Significant Sites | Ratio | p-Value | |
|---|---|---|---|---|
| Colorectal | 51 | 39 | 76% | 3.80 × 10−11 |
| Breast | 11 | 11 | 100% | 2.71 × 10−6 |
| Cervical | 7 | 7 | 100% | 2.87 × 10−4 |
| Lung | 16 | 15 | 94% | 2.91 × 10−7 |
| Total | 85 | 72 | 85% | 2.11 × 10−24 |
| % of Significant Sites | ||||
|---|---|---|---|---|
| Feature | #Sites | #Significant Sites | in Feature | in Total |
| North Shelf | 24,716 | 801 | 3.2% | 0.17% |
| North Shore | 62,647 | 2422 | 3.9% | 0.50% |
| CpG Island | 149,935 | 10,898 | 7.3% | 2.3% |
| South Shore | 49,055 | 1850 | 3.8% | 0.38% |
| South Shelf | 22,182 | 712 | 3.2% | 0.15% |
| Other | 172,655 | 6069 | 3.5% | 1.3% |
| Total | 481,190 | 22,752 | 4.7% | 4.7% |
| Horvath 2018 | Horvath 2013 | All Sites | |
|---|---|---|---|
| # sites | 391 | 353 | 481,190 |
| # significant sites | 338 | 334 | 22,752 |
| Mean of EFI | 1.29 | 1.26 | 1.18 |
| Std. of EFI | 0.33 | 0.34 | 0.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takenaka, Y.; Osaka Twin Research Group; Watanabe, M. Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins. Epigenomes 2024, 8, 44. https://doi.org/10.3390/epigenomes8040044
Takenaka Y, Osaka Twin Research Group, Watanabe M. Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins. Epigenomes. 2024; 8(4):44. https://doi.org/10.3390/epigenomes8040044
Chicago/Turabian StyleTakenaka, Yoichi, Osaka Twin Research Group, and Mikio Watanabe. 2024. "Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins" Epigenomes 8, no. 4: 44. https://doi.org/10.3390/epigenomes8040044
APA StyleTakenaka, Y., Osaka Twin Research Group, & Watanabe, M. (2024). Environmental Factor Index (EFI): A Novel Approach to Measure the Strength of Environmental Influence on DNA Methylation in Identical Twins. Epigenomes, 8(4), 44. https://doi.org/10.3390/epigenomes8040044