
The Phase Stability of Al3Er Studied by the First-Principles Calculations and Experimental Analysis

Abstract
:1. Introduction
2. Computation and Experiment Method
2.1. Computation Theory
2.2. Computational Details
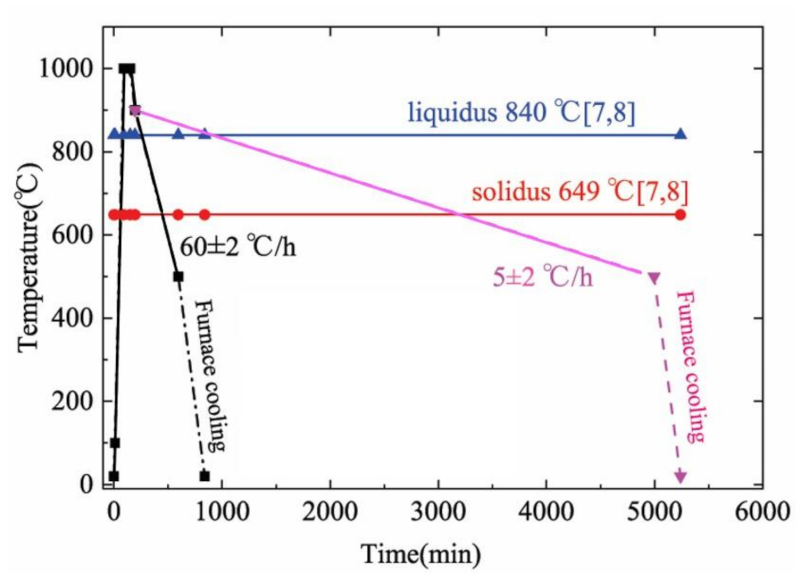
2.3. Experimental Method
3. Results
3.1. Computational Results
3.2. Experimental Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knipling, K.E.; Dunand, D.C.; Seidman, D.N. Criteria for developing castable, creep-resistant aluminum-based alloys—A review. Z. Fur Met. 2006, 97, 246–265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, K.; Wen, S.; Huang, H.; Nie, Z.; Zhou, D. The study on the coarsening process and precipitation strengthening of Al3Er precipitate in Al–Er binary alloy. J. Alloy. Compd. 2014, 610, 27–34. [Google Scholar] [CrossRef]
- Zhu, S.; Huang, H.; Nie, Z.; Wen, S.; Xing, Z. Investigation of the solubility of Er in aluminium. Light Met. 2009, 5, 018. [Google Scholar]
- Knipling, K.E.; Karnesky, R.A.; Lee, C.P.; Dunand, D.C.; Seidman, D.N. Precipitation evolution in Al–0.1Sc, Al–0.1Zr and Al–0.1Sc–0.1Zr (at.%) alloys during isochronal aging. Acta Mater. 2010, 58, 5184–5195. [Google Scholar] [CrossRef]
- Knipling, K. Precipitation evolution in Al–Zr and Al–Zr–Ti alloys during aging at 450–600 °C. Acta Mater. 2008, 56, 1182–1195. [Google Scholar]
- Xu, G.F.; Mou, S.Z.; Yang, J.J.; Nie, Z.; Yin, Z. Effect of trace rare earth element Er on Al-Zn-Mg alloy. Trans. Nonferrous Metals Soc. China 2006, 16, 598–603. [Google Scholar] [CrossRef]
- Okamoto, H. Al-Er (Aluminum-Erbium). J. Phase Equilibria Diffus. 2011, 32, 261–262. [Google Scholar] [CrossRef]
- Jin, L.; Kang, Y.-B.; Chartrand, P.; Fuerst, C.D. Thermodynamic evaluation and optimization of Al–Gd, Al–Tb, Al–Dy, Al–Ho and Al–Er systems using a Modified Quasichemical Model for the liquid. Calphad 2011, 34, 456–466. [Google Scholar] [CrossRef]
- Cacciamani, G.; Saccone, A.; De Negri, S.; Ferro, R. The Al-Er-Mg Ternary SystemPart I Thermodynamic Modeling. J. Phase Equilibria 2002, 23, 29–37. [Google Scholar] [CrossRef]
- Cacciamani, G.; Saccone, A.; De Negri, S.; Ferro, R. The Al-Er-Mg Ternary SystemPart II Thermodynamic Modeling. J. Phase Equilibria 2002, 23, 38–50. [Google Scholar] [CrossRef]
- Meyer, A. Über die kubische und rhomboedrische form des Al3Er. J. Less Common Met. 1970, 20, 353–358. [Google Scholar] [CrossRef]
- van Vucht, J.H.N.; Buschow, K.H.J. The structures of the rare-earth trialuminides. J. Less Common Met. 1966, 10, 98–107. [Google Scholar] [CrossRef]
- Gao, M.C.; Rollett, A.D.; Widom, M. Lattice stability of aluminum-rare earth binary systems: A first-principles approach. Phys. Rev. B 2007, 75, 4120. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Seidman, D.N.; Wolverton, C. First-principles phase stability, magnetic properties and solubility in aluminum–rare-earth (Al–RE) alloys and compounds. Acta Mater. 2011, 59, 3659–3666. [Google Scholar] [CrossRef]
- Glensk, A.; Grabowski, B.; Hickel, T.; Neugebauer, J. Understanding Anharmonicity in fcc Materials: From its Origin to ab initio strategies beyond the Quasiharmonic Approximation. Phys. Rev. Lett. 2005, 114, 195901. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Kavakbasi, B.T.; Dutta, B.; Grabowski, B.; Peterlechner, M.; Hickel, T.; Divinski, S.V.; Wilde, G.; Neugebauer, J. Low-temperature features in the heat capacity of unary metals and intermetallics for the example of bulk aluminum andAl3Sc. Phys. Rev. B 2017, 95, 094307. [Google Scholar] [CrossRef] [Green Version]
- Kohan, A.F.; Tepesch, P.D.; Ceder, G.; Wolverton, C. Computation of alloy phase diagrams at low temperatures. Comput. Mater. Sci. 1998, 9, 389–396. [Google Scholar] [CrossRef]
- Wolverton, C. Crystal structure and stability of complex precipitate phases in Al–Cu–Mg–(Si) and Al–Zn–Mg alloys. Acta Mater. 2001, 49, 3129–3142. [Google Scholar] [CrossRef]
- Wolverton, C. First-principles theory of 250,000-atom coherent alloy microstructure. Model. Simul. Mater. Sci. Eng. 2000, 8, 323–333. [Google Scholar] [CrossRef]
- Aidhy, D.S.; Zhang, Y.S.; Wolverton, C. Prediction of a Ca(BH4)(NH2) quaternary hydrogen storage compound from first-princi ples calculations. Phys. Rev. B 2011, 84, 134103. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, S.S.; Hegde, V.I.; Saboo, A.; Wolverton, C. Energetics of cobalt alloys and compounds and solute–vacancy binding in fcc cobalt: A first-principles database. Acta Mater. 2017, 124, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wolverton, C.; Ozolins, V. First-principles aluminum database: Energetics of binary Al alloys and compounds. Phys. Rev. B 2006, 73, 4104. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B Condens Matter. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B Condens Matter. 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B Condens Matter. 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Blochl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B Condens Matter. 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Bargouth, M.O.; Will, G. The magnetic structure of hexagonal ErAl3. Phys. Lett. A 1971, 36, 50–51. [Google Scholar] [CrossRef]
- Bargouth, M.O.; Will, G.; Yelon, W.B.; Meyer, A. Neutron Crystal-Field Spectroscopy in Cubic ErAl3. J. Phys. C-Solid State Phys. 1976, 9, 1483–1489. [Google Scholar] [CrossRef]
- van Pieterson, L.; Reid, M.F.; Burdick, G.W.; Meijerink, A. 4fn→4fn−15d transitions of the heavy lanthanides: Experiment and theory. Phys. Rev. B 2002, 65, 045114. [Google Scholar] [CrossRef]
- Duan, C.G.; Komesu, T.; Jeong, H.K.; Borca, C.N.; Yin, W.G.; Liu, J.; Mei, W.N.; Dowben, P.A.; Petukhov, A.G.; Schultz, B.D.; et al. Hybridization between 4f-5d states in ErAs(100). Surf. Rev. Lett. 2004, 11, 531–539. [Google Scholar] [CrossRef]
- Losovyj, Y.B.; Tang, J.; Wang, W.; Hong, Y.; Palshin, V.; Tittsworth, R. 4f–5d hybridization in a high k dielectric. Phys. Lett. A 2006, 357, 240–244. [Google Scholar] [CrossRef]
- Brown, S.D.; Strange, P.; Bouchenoire, L.; Thompson, P.B.J. 4f/5d hybridization in the heavy rare earth elements Er and Tm. Phys. Rev. B 2013, 87, 5111. [Google Scholar] [CrossRef] [Green Version]
- Kirklin, S.; Saal, J.E.; Hegde, V.I.; Wolverton, C. High-throughput computational search for strengthening precipitates in alloys. Acta Mater. 2016, 102, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Issa, A.; Saal, J.E.; Wolverton, C. Physical factors controlling the observed high-strength precipitate morphology in Mg–rare earth alloys. Acta Mater. 2014, 65, 240–250. [Google Scholar] [CrossRef]
- Issa, A.; Saal, J.E.; Wolverton, C. Formation of high-strength β′ precipitates in Mg–RE alloys: The role of the Mg/β″ interfacial instability. Acta Mater. 2015, 83, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Chaput, L.; Tanaka, I. Distributions of phonon lifetimes in Brillouin zones. Phys. Rev. B 2015, 91, 094306. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, J.L.; Gordon, R.O.; Humphreys, J.E. Some new intermetallic compounds of holmium and erbium with Ag, Au, Pt, Al, In, Tl, and Ge. Acta Crystallogr. 1965, 19, 285–286. [Google Scholar] [CrossRef]
- Cannon, J.F.; Hall, H.T. Effect of high pressure on the crystal structures of lanthanide trialuminides. J. Less Common Met. 1975, 40, 313–328. [Google Scholar] [CrossRef]
- Tao, X.; Ouyang, Y.; Liu, H.; Feng, Y.; Du, Y.; Jin, Z. First-principles calculations of the thermodynamic and elastic properties of the L12-based Al3RE (RE = Sc, Y., La–Lu). Int. J. Mater. Res. 2008, 99, 582–588. [Google Scholar] [CrossRef]

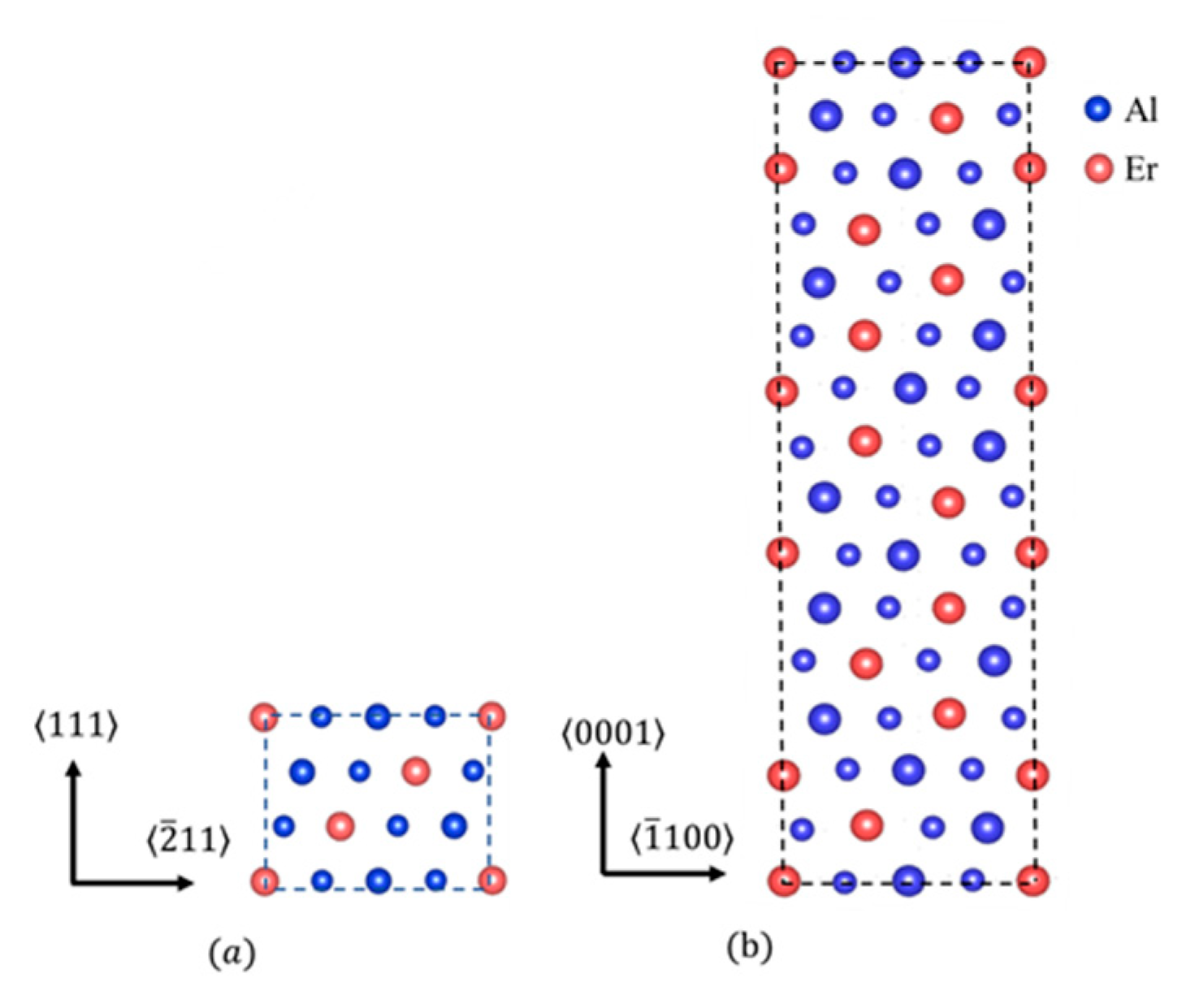

{kind=link}
{kind=link}
{kind=link}
| Structure | Lattice Constants/ Å | Difference/% | |||
|---|---|---|---|---|---|
| ST | FC | EXP | ST | FT | |
| Cu3Au.cP4 | 4.215 | 4.238 | 4.215 [12] | 0.0 0.1 0.0 | 0.5 |
| 4.212 0.002 [42] | 0.6 | ||||
| 4.214 [43] | 0.6 | ||||
| Al3Ho.hR20 | 5.993 | 6.063 | 6.025 [12] | −0.5 | 0.6 |
| 5.993 | 6.063 | 6.025 [12] | |||
| 36.059 | 35.820 | 35.675 [12] | 1.1 | 0.4 | |
| Structure | Formation Energy with ST | Formation Energy with FC | Relative Energy | Ref. | |
|---|---|---|---|---|---|
| ST | FC | ||||
| eV/atom | |||||
| Al3Ho.hR20 | −0.412 | −0.417 * | 0 | 0 * | |
| −0.412 ** | 0 ** | ||||
| Cu3Au.cP4 | −0.411 | −0.416 * | 0.001 | 0.001 * | −0.416 [13] −0.416 [39] −0.417 [44] |
| −0.404 ** | 0.008 ** | ||||
| Al3Gd.hR12 | −0.400 | −0.413 * | 0.012 | 0.004 * | |
| AlNd3.hP8 | −0.399 | −0.345 * | 0.013 | 0.072 * | |
| Ni3Ti.hP16 | −0.342 | −0.345 * | 0.060 | 0.072 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Gao, K.; Ding, Y.; Li, H.; Wu, X.; Wen, S.; Gao, M.; Huang, H.; Nie, Z.; Zhou, D. The Phase Stability of Al3Er Studied by the First-Principles Calculations and Experimental Analysis. Metals 2021, 11, 759. https://doi.org/10.3390/met11050759
Gao C, Gao K, Ding Y, Li H, Wu X, Wen S, Gao M, Huang H, Nie Z, Zhou D. The Phase Stability of Al3Er Studied by the First-Principles Calculations and Experimental Analysis. Metals. 2021; 11(5):759. https://doi.org/10.3390/met11050759
Chicago/Turabian StyleGao, Chunlai, Kunyuan Gao, Yusheng Ding, Haonan Li, Xiaolan Wu, Shengping Wen, Mu Gao, Hui Huang, Zuoren Nie, and Dejing Zhou. 2021. "The Phase Stability of Al3Er Studied by the First-Principles Calculations and Experimental Analysis" Metals 11, no. 5: 759. https://doi.org/10.3390/met11050759