
A First-Principles Study on Na and O Adsorption Behaviors on Mo (110) Surface


Abstract
:1. Introduction
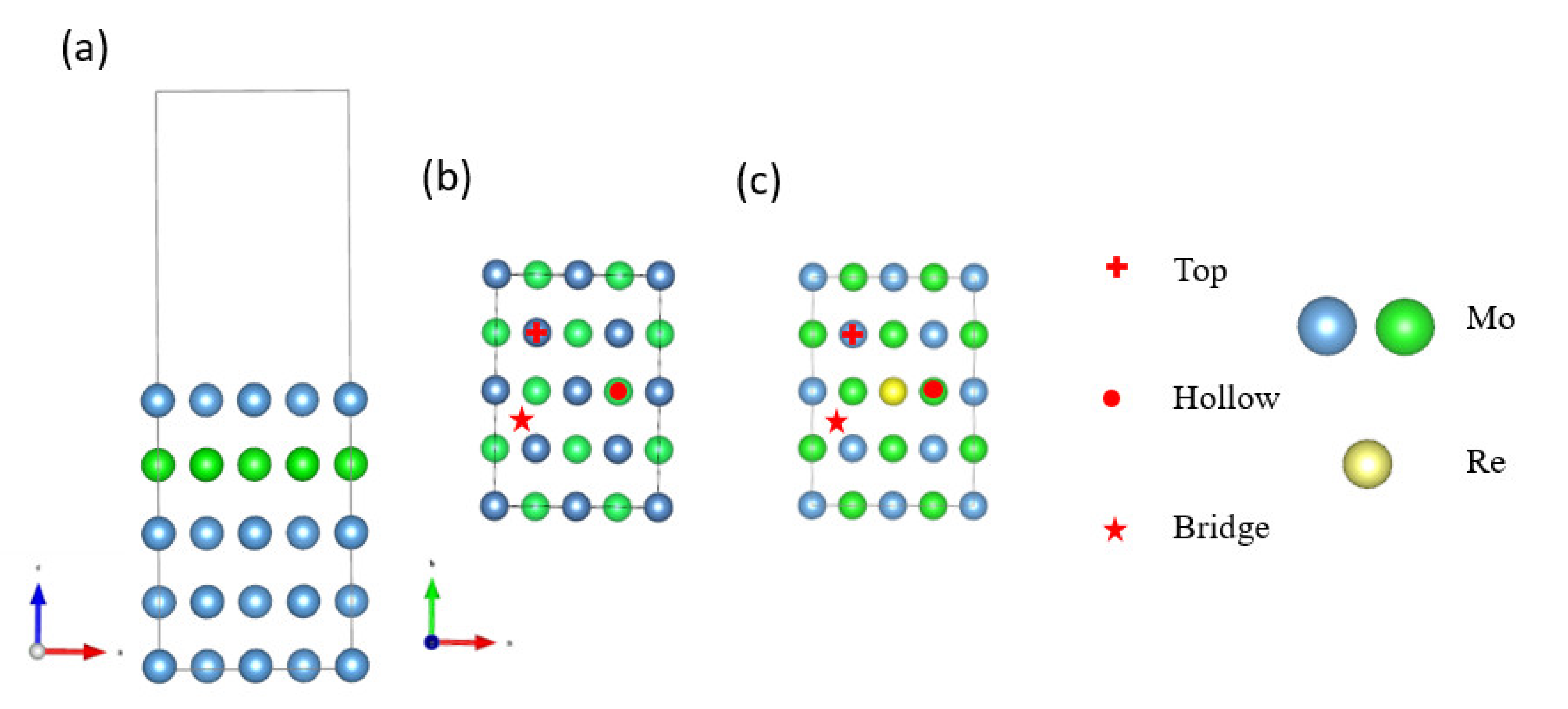
2. Computational Methods and Model
3. Results and Discussion
3.1. Model Verification
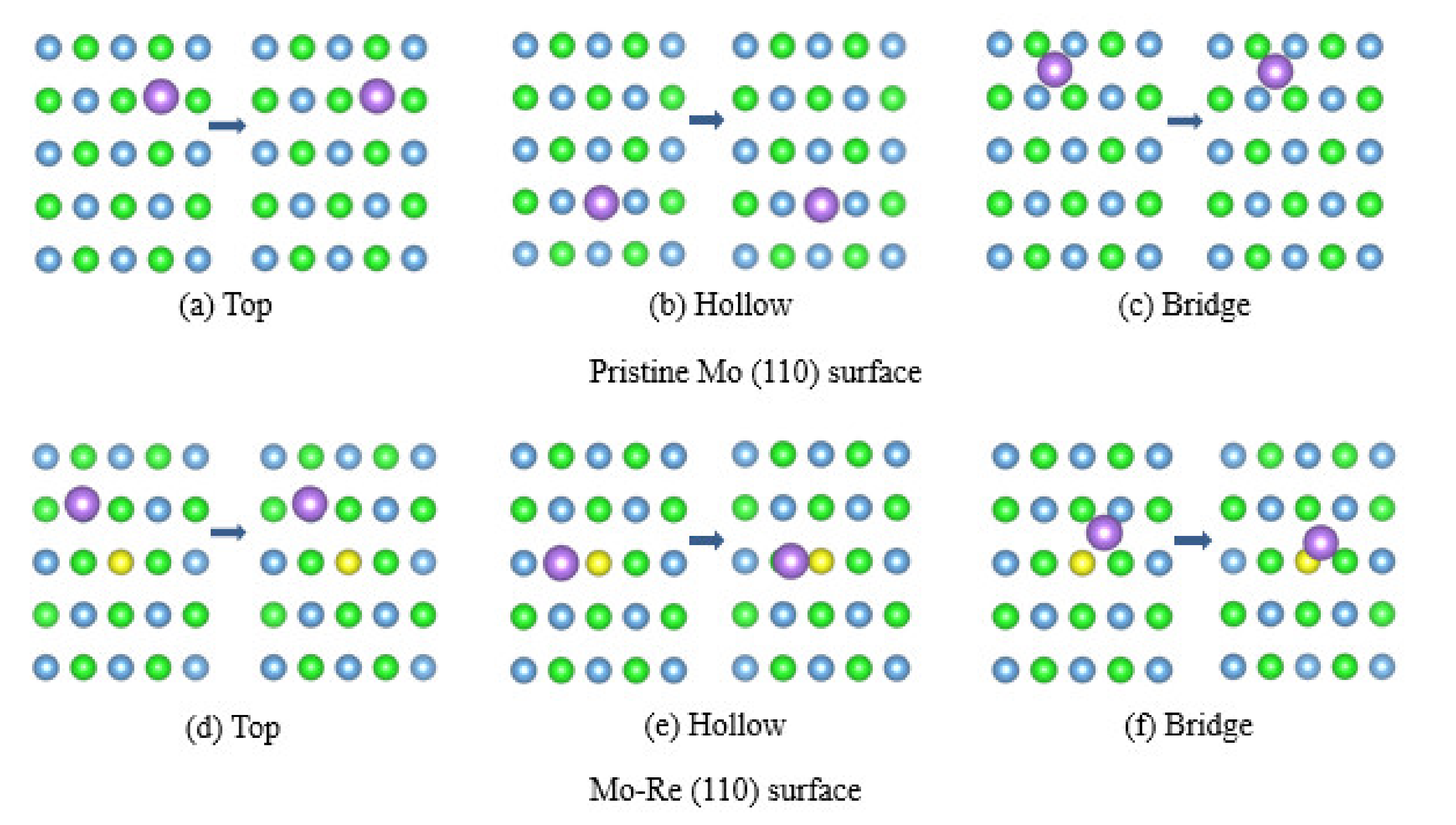
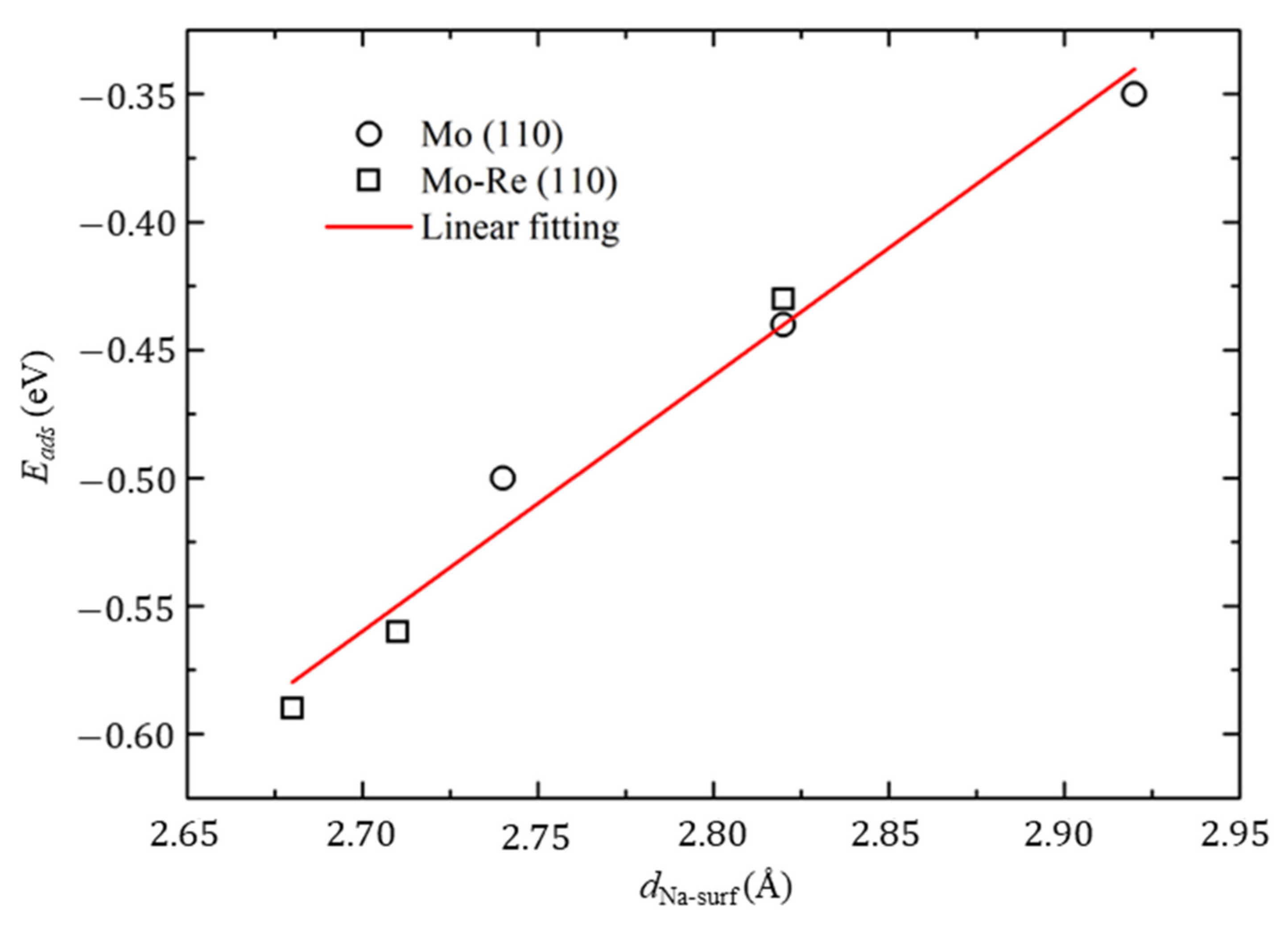
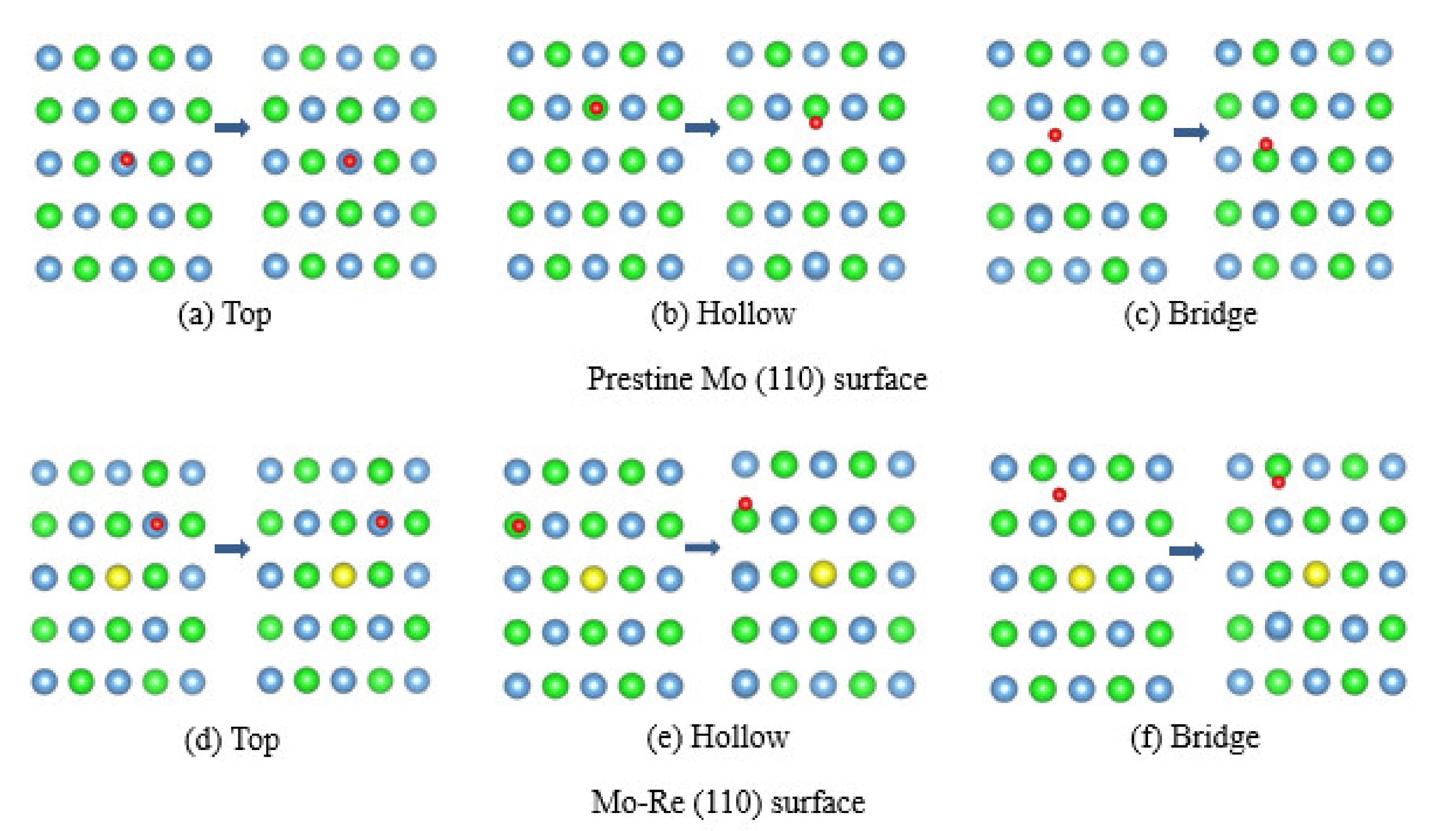
3.2. Na Atom and O Atom Adsorption
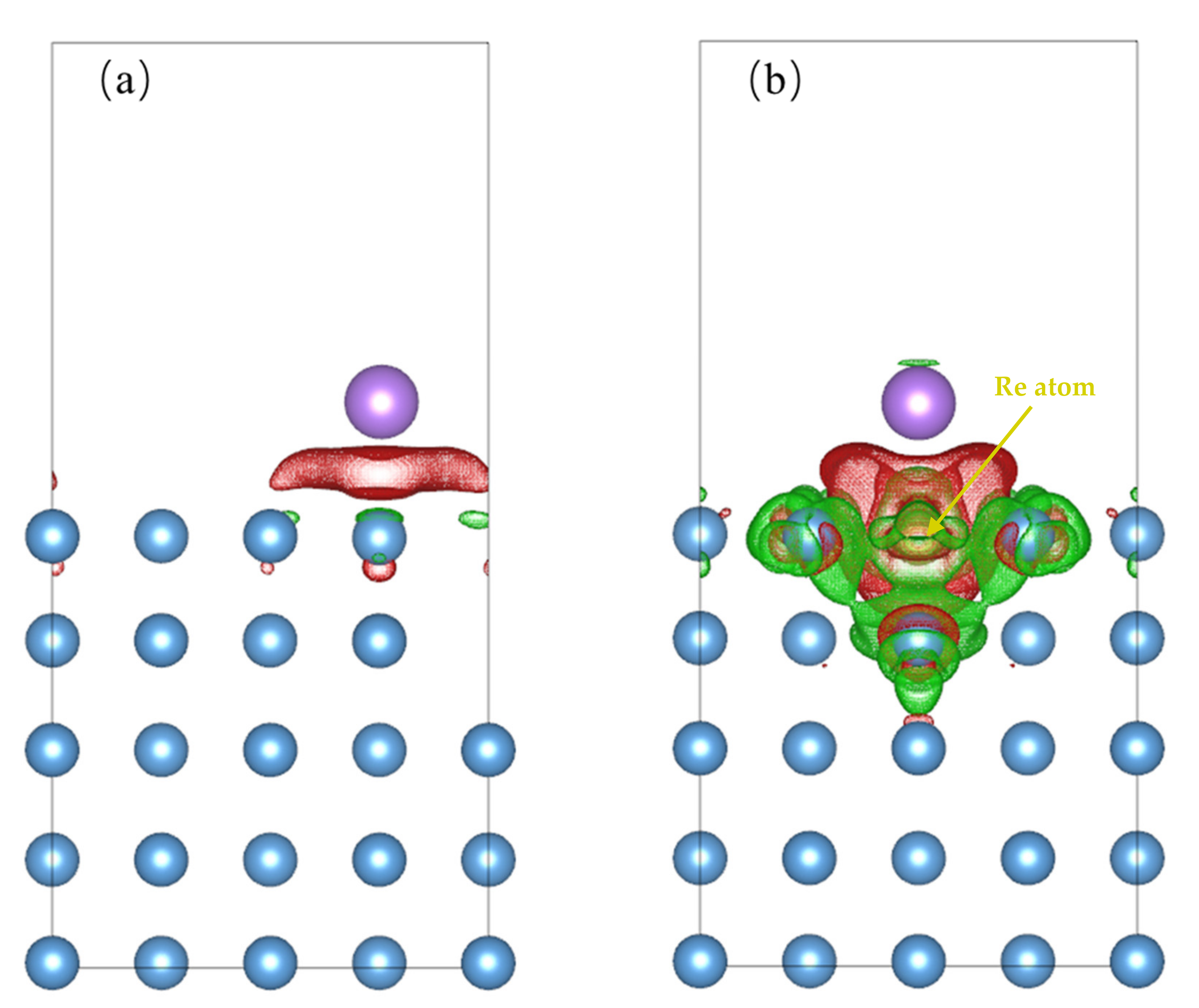
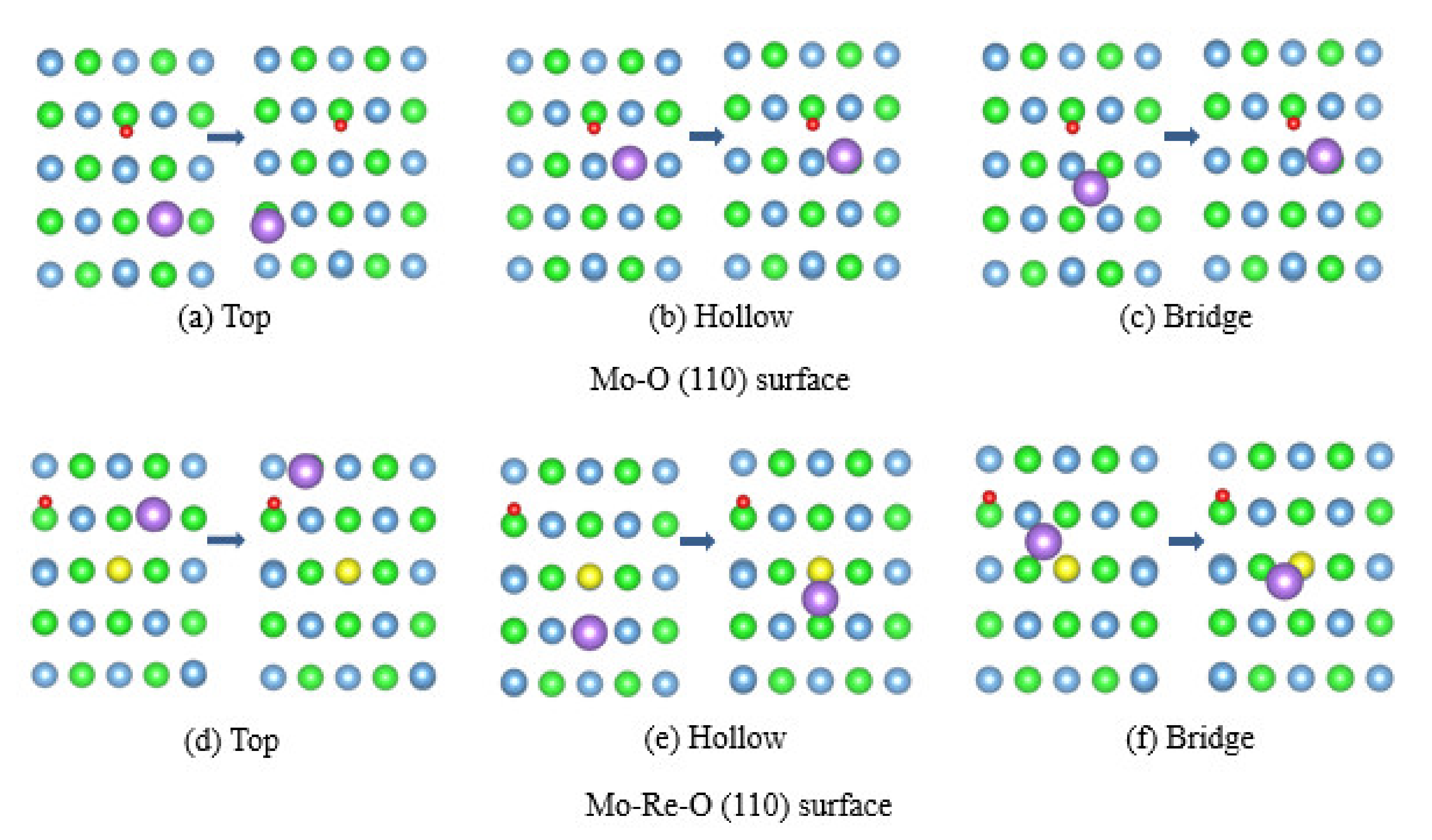
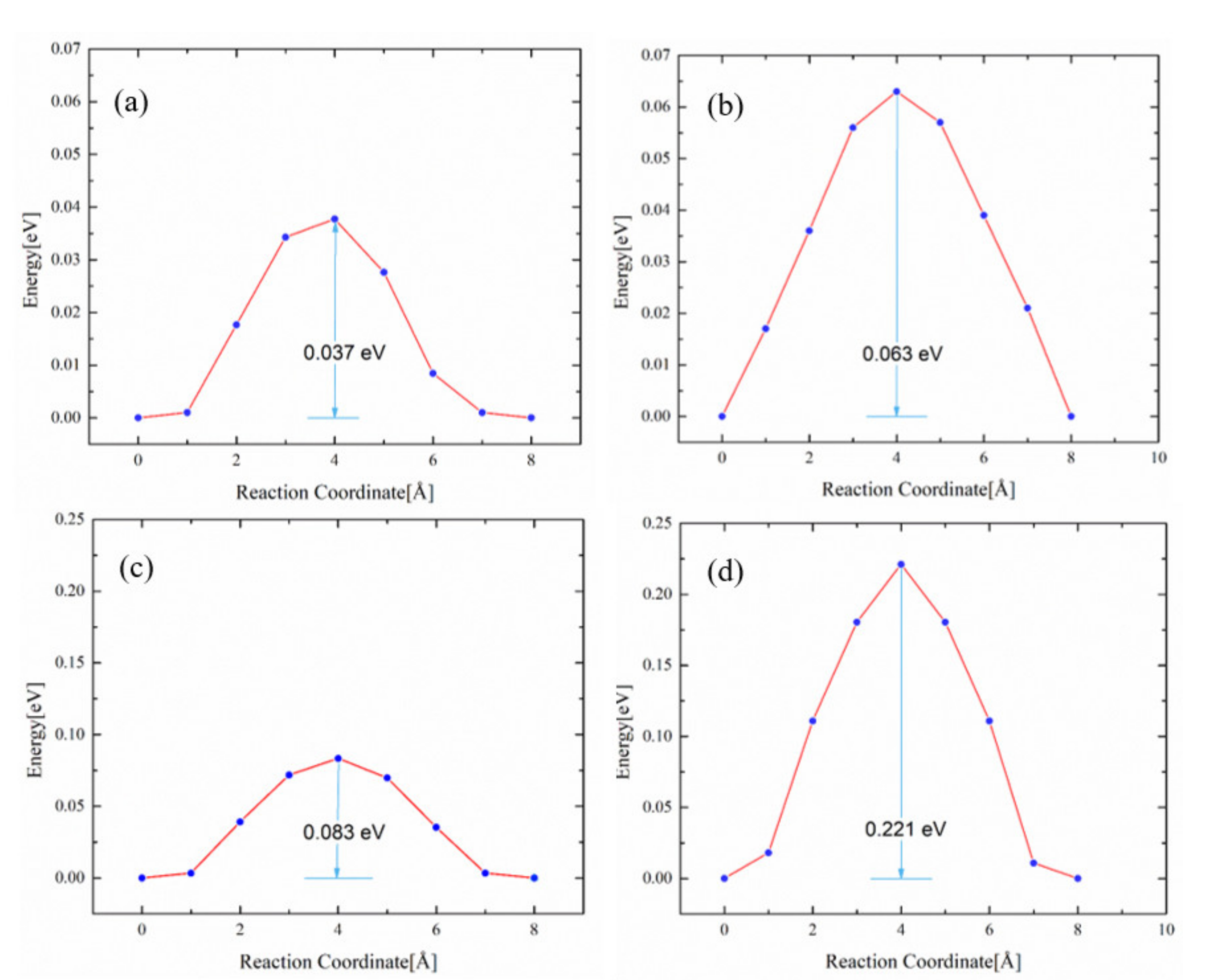
3.3. Impact of O on Na Adsorption and Diffusion
3.4. Impact of O/Na atom Adsorption on the Vacancy Formation of Mo (110) and Mo-Re (110) Surfaces
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Zhuang, J. Research, development and industrial application of heat pipe technology in China. Appl. Therm. Eng. 2003, 23, 1067–1083. [Google Scholar] [CrossRef]
- Ai, B.; Chen, S.; Yu, J.; Lu, Q.; Han, H.; Hu, L. Fabrication of lithium/C-103 alloy heat pipes for sharp leading edge cooling. Heat Mass Transf. 2017, 54, 1359–1366. [Google Scholar] [CrossRef]
- Wang, C.; Guo, Z.; Zhang, D.; Qiu, S.; Tian, W.; Wu, Y.; Su, G. Transient behavior of the sodium–potassium alloy heat pipe in passive residual heat removal system of molten salt reactor. Prog. Nucl. Energy 2013, 68, 142–152. [Google Scholar] [CrossRef]
- Faghri, A. Review and Advances in Heat Pipe Science and Technology. J. Heat Transf. 2012, 134, 123001. [Google Scholar] [CrossRef]
- Reay, D.; McGlen, R.; Kew, P. Heat Pipes: Theory, Design and Applications, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Jouhara, H.; Chauhan, A.; Nannou, T.; Almahmoud, S.; Delpech, B.; Wrobel, L. Heat pipe based systems—Advances and applications. Energy 2017, 128, 729–754. [Google Scholar] [CrossRef]
- Qu, W. Progress Works of High and Super High Temperature Heat Pipes. In Developments in Heat Transfer; Bernardes, M.A.D.S., Ed.; InTech: London, UK, 2011; pp. 503–522. ISBN 978-953-307-569-3. [Google Scholar]
- El-Genk, M.; Tournier, J.-M. Challenges and fundamentals of modeling heat pipes’ startup from a frozen state. AIP Conf. Proc. 2002, 608, 127–138. [Google Scholar]
- Lundberg, L.B. Refractory Metals in Space Nuclear Power. JOM 1985, 37, 44–47. [Google Scholar] [CrossRef]
- Lundberg, L. An evaluation of molybdenum and its alloys. In Proceedings of the 16th Thermophysics Conference, American Institute of Aeronautics and Astronautics (AIAA), Palo Alto, CA, USA, 23–25 June 1981. [Google Scholar]
- King, J.C.; El-Genk, M.S. Review of Refractory Materials for Alkali Metal Thermal-to-Electric Conversion Cells. J. Propuls. Power 2001, 17, 547–556. [Google Scholar] [CrossRef]
- El-Genk, M.S.; Tournier, J.-M. A review of refractory metal alloys and mechanically alloyed-oxide dispersion strengthened steels for space nuclear power systems. J. Nucl. Mater. 2005, 340, 93–112. [Google Scholar] [CrossRef]
- Hampel, C. Refractory Metals. Tantalum, Niobium, Molybdenum, Rhenium, and Tungsten. Ind. Eng. Chem. 1961, 53, 90–96. [Google Scholar] [CrossRef]
- Shmelev, A.N.; Kozhahmet, B.K. Use of molybdenum as a structural material of fuel elements for improving the safety of nuclear reactors. J. Phys. Conf. Ser. 2017, 781, 012022. [Google Scholar] [CrossRef]
- Gilbert, M.; Packer, L.; Stainer, T. Experimental validation of inventory simulations on molybdenum and its isotopes for fusion applications. Nucl. Fusion 2020, 60, 106022. [Google Scholar] [CrossRef]
- Lundberg, L. Critical Evaluation of Molybdenum and Its Alloys for Use in Space Reactor Core Heat Pipes; No. LA-8685-MS; Los Alamos Scientific Laboratory: Los Alamos, NM, USA, 1981. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Kumar, K. Uniaxial, load-controlled cyclic deformation of recrystallized molybdenum sheet. Mater. Sci. Eng. A 2012, 540, 187–197. [Google Scholar] [CrossRef]
- Jörg, T.; Music, D.; Hauser, F.; Cordill, M.J.; Franz, R.; Köstenbauer, H.; Winkler, J.; Schneider, J.; Mitterer, C. Deformation behavior of Re alloyed Mo thin films on flexible substrates: In situ fragmentation analysis supported by first-principles calculations. Sci. Rep. 2017, 7, 7374. [Google Scholar] [CrossRef]
- Agnew, S.R.; Leonhardt, T. The low-temperature mechanical behavior of molybdenum-rhenium. JOM 2003, 55, 25–29. [Google Scholar] [CrossRef]
- DiStefano, J.R. Review of alkali metal and refractory alloy compatibility for Rankine cycle applications. J. Mater. Eng. 1989, 11, 215–225. [Google Scholar] [CrossRef]
- Tu, S.-T.; Zhang, H.; Zhou, W.W. Corrosion failures of high temperature heat pipes. Eng. Fail. Anal. 1999, 6, 363–370. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, A.J. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. J. Rev. Mod. Phys. 1992, 64, 1045. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H. Supplemental Literature Review of Binary Phase Diagrams: Ag-Yb, Al-Co, Al-I, Co-Cr, Cs-Te, In-Sr, Mg-Tl, Mn-Pd, Mo-O, Mo-Re, Ni-Os, and V-Zr. J. Phase Equilibria Diffus. 2016, 37, 726–737. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- American Society for Metals and ASM Handbook Committee. Properties and Selection: Nonferrous Alloys and Pure Metals, 9th ed.; American Society for Metals: Russell Township, OH, USA, 1979. [Google Scholar]
- Guo, F.; Wang, J.; Du, Y.; Wang, J.; Shang, S.-L.; Li, S.; Chen, L. First-principles study of adsorption and diffusion of oxygen on surfaces of TiN, ZrN and HfN. Appl. Surf. Sci. 2018, 452, 457–462. [Google Scholar] [CrossRef]
- Graciani, J.; Sanz, J.F.; Asaki, T.; Nakamura, K.; Rodriguez, J.A. Interaction of oxygen with TiN(001):N⟷O exchange and oxidation process. J. Chem. Phys. 2007, 126, 244713. [Google Scholar] [CrossRef] [PubMed]
- Osei-Agyemang, E.; Balasubramanian, G. Surface oxidation mechanism of a refractory high-entropy alloy. Npj Mater. Degrad. 2019, 3, 20. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Energy(eV/Å2) | ||
|---|---|---|
| Surface | Present Calculation | Materials Project Database |
| (100) | 0.22 | 0.22 |
| (110) | 0.17 | 0.17 |
| (111) | 0.18 | 0.18 |
| Surface | Initial Site | Final Site | ||
|---|---|---|---|---|
| Mo (110) | Top | 2.92 | −0.35 | Top |
| Hollow | 2.74 | −0.51 | Hollow | |
| Bridge | 2.82 | −0.44 | Bridge | |
| Mo-Re (110) | Top | 2.82 | −0.43 | Top |
| Hollow | 2.71 | −0.56 | Hollow | |
| Bridge | 2.68 | −0.60 | Bridge |
| Surface | Initial Site | Final Site | ||
|---|---|---|---|---|
| Mo (110) | Top | 2.18 | −2.80 | Top |
| Hollow | 1.15 | −4.09 | Hollow | |
| Bridge | 1.15 | −4.09 | Hollow | |
| Top | 1.73 | −2.82 | Top | |
| Mo-Re (110) | Hollow | 1.14 | −4.14 | Hollow |
| Bridge | 1.14 | −4.14 | Hollow |
| Surface | Initial Site | Final Stie | |||
|---|---|---|---|---|---|
| Mo-O (110) | Top | 6.02 | 2.78 | −0.52 | Hollow |
| Hollow | 2.36 | 2.68 | −0.53 | Hollow | |
| Bridge | 2.35 | 2.83 | −0.52 | Hollow | |
| Mo-Re-O (110) | Top | 2.37 | 2.76 | −0.54 | Hollow |
| Hollow | 4.77 | 2.71 | −0.61 | Hollow | |
| Bridge | 3.18 | 2.77 | −0.57 | Bridge |
| Adsorbate | Surface | Vacancy Formation Energy (eV) |
|---|---|---|
| Clean | Mo (110) | 1.45 |
| Mo-Re (110) | 1.32 | |
| A single O atom | Mo (110) | 0.97 |
| Mo-Re (110) | 0.86 | |
| A single Na atom | Mo (110) | 0.93 |
| Mo-Re (110) | 0.90 | |
| Na and O co-adsorption | Mo (110) | 0.47 |
| Mo-Re (110) | 1.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Q.; Liu, Z.; Liang, W.; Ma, M.; Deng, H. A First-Principles Study on Na and O Adsorption Behaviors on Mo (110) Surface. Metals 2021, 11, 1322. https://doi.org/10.3390/met11081322
Zeng Q, Liu Z, Liang W, Ma M, Deng H. A First-Principles Study on Na and O Adsorption Behaviors on Mo (110) Surface. Metals. 2021; 11(8):1322. https://doi.org/10.3390/met11081322
Chicago/Turabian StyleZeng, Qingqing, Zhixiao Liu, Wenfeng Liang, Mingyang Ma, and Huiqiu Deng. 2021. "A First-Principles Study on Na and O Adsorption Behaviors on Mo (110) Surface" Metals 11, no. 8: 1322. https://doi.org/10.3390/met11081322
APA StyleZeng, Q., Liu, Z., Liang, W., Ma, M., & Deng, H. (2021). A First-Principles Study on Na and O Adsorption Behaviors on Mo (110) Surface. Metals, 11(8), 1322. https://doi.org/10.3390/met11081322