Abstract
Segregation of foreign atoms on a solid substrate in a liquid metal modifies the geometry and chemistry of the substrate surface and, correspondingly, its potency to nucleate a solid metal. We here investigate the effects of the segregation of alkaline earth (AE) atoms, Mg, Sr, and Ba, at the interfaces between liquid Al and γ-Al2O3{1 1 1} substrates using an ab initio molecular dynamics method. This study reveals the high stability and localized nature of the segregated AE atoms at the oxide substrates. The segregation of the AE atoms induces reconstruction of the metal atoms terminating the oxide substrates, and causes atomic roughness of the substrate surfaces. The content of the induced atomic roughness relates to the ionic size of the AE atoms. Correspondingly, the potency of the oxide substrates is modified. This indicates the possibility of manipulating the substrate potency via segregation of selected impure atoms, which would help to control solidification processes.
1. Introduction
Solid particles in liquids reduce the barrier of nucleation and catalyze the phase transition from liquid to solid, which is referred to as heterogeneous nucleation [1,2,3]. Heterogeneous nucleation, thus, occurs most often, while homogeneous nucleation in liquid without heterogeneous particles happens rarely [3,4]. Heterogeneous nucleation is determined by the potency of the substrate to the metal. The more potent the substrate, the less driving force is required [3,4,5]. In metallurgy, the foreign particles are referred to as grain refiners.
In order to obtain cast parts and metals/alloys of fine and uniform microstructures and desirable mechanical performances, grain refiners were developed [2,3,4,6] and have been widely used for the purpose of casting light metals. At present, there are two strategies to reach fine and uniform microstructures in cast parts and alloys [3,4,5]. The conventional approach is to search for substrates of maximum potency to the metal, via segregation of impurities [3,4,5,6,7]. A successful case is the application of the Al-nTi-B (n = 3, 5) grain refiners for casting Al-based metals [2,3,4,5,6,7]. Transmission electron microscopy (TEM) observations discovered that segregation of Ti atoms causes the formation of a Ti-rich two-dimensional compound, most likely TiAl3, at the Ti-terminated TiB2{0 0 0 1} surface [8]. The two-dimensional compound maximizes the potency of this solid substrate to nucleate solid Al [4,8]. When the grain initiation temperature is reached, grain initiation occurs first at the largest particles, and gradually at smaller ones [3,9]. This process is called progressive grain initiation (PGI) [4,5], during which only a small fraction of particles function as grain initiation sites.
The early-stage solidification scheme [4,5] also suggests another approach to produce cast parts/alloys of fine and uniform microstructures via using substrates of minimized potency. A substrate of minimum potency requires a large driving force in order to nucleate a solid metal phase so that the nucleation temperature may be close to or even lower than that of the grain initiation temperature. In such cases, as the grain initiation temperature is reached, nucleation and grain initiation occur in small time intervals. Thus, grain initiation occurs in an explosive way, which is thus referred to as explosive grain initiation (EGI). In such processes, a large fraction of particles, even those with small sizes, can act as nucleation sites. Naturally, in both PGI and EGI processes, densely and uniformly distributed particles of similar potency are vital to achieve grain refinement for the cast parts and alloys.
During liquid dealing and casting of Al-based metals and alloys, γ-Al2O3 particles are formed unavoidably [10,11,12,13]. TEM analysis disclosed that the newly formed γ-Al2O3 particles exhibit dominantly plate-shaped morphologies with the {1 1 1} facets [10,11,13]. These native oxide particles have impacts not only on the mechanical performances of the cast parts and/or alloys, but also may act as potential nucleation sites during solidification [4].
The crystal structure of γ-Al2O3 was experimentally determined to be a defective spinel-type structure [14,15]. Along its [1 1 1] axis, the structure of γ-Al2O3 is composed alternately of an Al layer (a single Al layer or three sublayers) and an oxygen layer [14,15]. The oxygen layers are composed of distorted two-dimensional hexagonal networks. Recent ab initio molecular dynamics simulations disclosed that the liquid Al atoms adjacent to the substrates exhibit atomic ordering at the interfaces between liquid Al and γ-Al2O3{1 1 1} substrate surfaces, which are hereafter denoted as Al(l)/γ-Al2O3{1 1 1} interfaces [16]. There are two kinds of geometrically different Al layers terminating the oxide substrates. One is terminated by a single Al atomic layer (L-Al/γ-Al2O3{1 1 1}Al2) with a statistically flat Al layer (S2 in short). Another one is terminated by three Al sublayers (denoted as L-Al/γ-Al2O3{1 1 1}Al1, S1), which are atomically rough.
The atomic ordering in liquid bordering to a solid substrate above the nucleation temperature is referred to as prenucleation. Prenucleation at a liquid/solid interface contributes a predecessor for following heterogeneous nucleation in the liquid [4,17,18,19,20]. Thus, prenucleation at a liquid/solid interface bonds to the potency of the substrate to nucleate a solid. From the recent ab initio molecular dynamics (AIMD) study [16], it was found that the potencies of the two L-Al/γ-Al2O3{1 1 1} interfaces are different from each other. Moreover, the AIMD simulations produced similar total-valence-electron energies for the two different interfaces, implying the coexistence of the two oxide substrates in liquid Al metals. The different potencies implies that only part of the particles may behave as potential nucleation sites. The density of potential nucleation sites will be increased if we can modify the substrate surfaces to make two substrate surfaces attain similar potencies by means of segregation of impurities.
As mentioned earlier, segregation of foreign atoms on substrates has been long utilized to manipulate the potency of the substrates to nucleate a metal. This is exampled by the segregation of extra Ti atoms on the Ti-terminated TiB2{0 0 0 1} surface in liquid Al to enhance its potency, by which fine and uniform microstructures in the cast parts have been achieved [6,7,8,9]. There are also opposite cases, e.g., segregation of Zr or Si atoms at the TiB2{0 0 0 1} substrates to poison the grain refiners [21,22,23]. Ab initio molecular dynamics simulations uncovered that the segregated Zr atoms cause atomic roughness at the Ti-terminated TiB2{0 0 0 1} substrate surfaces [22]. Consequently, the prenucleation content at the Zr-segregated L-Al/TiB2{0 0 0 1} interface becomes deteriorated.
Additionally, in the study of grain refinements of MgO particles in Mg-0.5Ca alloys, Wang, et al. observed adsorption layers of impurity Al, Ca and N atoms on the MgO{1 1 1} facets [24]. Ma, et al. observed Cu segregation at the Al/α-Al2O3 interfaces [25]. Recently, segregation of the nd1 elements, La or Y, at the Al/γ-Al2O3{1 1 1} interfaces has been realized experimentally and observed by TEM techniques [26]. Meanwhile, theoretical approaches, especially parameter-free AIMD methods, have been employed to investigate the atomic ordering at interfaces between liquid metal and oxide substrates [16,27,28,29]. Recent AIMD modeling also discovered that the segregated nd1 atoms are bonded with the outmost O ions, and exhibit localized characteristics. Consequently, the substrate surfaces grow to become rougher [30]. The segregation of the nd1 atoms thus reduces prenucleation at the interfaces.
In order to obtain comprehensive knowledge about the impact of segregation of impurities of different chemical natures on prenucleation at the interfaces between liquid metal and solid substrates, we investigated several groups of elements of lower electronegativity values than Al, including alkaline earth (AE) elements, for segregation at the propotype Al/γ-Al2O3{1 1 1} interfaces.
Alkaline earth atoms, including Mg, Sr, and Ba, exist in many Al metals and alloys [11,12,13]. They have smaller electronegativity values than that of Al. Thus, they may segregate at the L-Al/γ-Al2O3{1 1 1} interfaces. Moreover, the AE atoms have two s, p valence electrons, making them different from Al atoms, which have three. The ionic/atomic size of the alkaline earth elements increases from Mg to Sr to Ba. The AE atoms contain only two s, p valence electrons, which is also different from the Sc, Y, and L atoms, which have three valence electrons, including one d-electron. This provides an ideal case for us to investigate both size and valence effects on the prenucleation of impurity segregations at a liquid metal/oxide interface.
We here investigate the effect of segregation of the AE atoms Mg, Sr, and Ba at the L-Al/γ-Al2O3{1 1 1} interfaces, systematically employing ab initio molecular dynamics simulation techniques. The simulations disclose the high stability and localization of the segregated AE atoms that are bonded to the O atoms at the substrates. The segregation causes atomic roughness of the substrate surfaces, which impacts the potency of the substrates. The obtained information is also useful for improving our understanding of prenucleation at interfaces between liquid light metals and other oxide substrates, including liquid-Al/α-Al2O3 [27,31,32,33,34,35], liquid-Y/α-Al2O3 [29], solid-Cu/α-Al2O3 [36], and others [28,37,38].
2. Details of Methods
In the present study, we adopted a reverse approach based on our previous research [16,30]. We used the experimental structure of γ-Al2O3 in the literature [14,15] to set up the oxide substrates. The built L-Al/γ-alumina interface systems have a hexagonal supercell with a = a0, where a0 is the lattice parameter of the cubic γ-Al2O3 phase at the simulation temperature [14,15,16]. The length of the c-axis was obtained according to the thickness of the oxide slab and the atomic volumes of the liquid metals. We thus obtained a hexagonal cell with a = 11.37 Å, c = 37.18 Å. This cell contained 310 atoms: 96 O and 214 (Al + AE) atoms. There were four AE atoms on each surface, which corresponded to about 5at.% of AE in the Al liquid. Test of systems with different c-axis lengths showed no significant influences on the results.
We used a pseudo-potential plane-wave method in the first-principles code VASP [39] for the first-principles calculations and AIMD simulations. This first-principles code permits variable fractional occupation numbers. It worked well for insulator/metal interfaces [16,30,37,38,39]. It uses finite-temperature density functional theory, the exact energy minimization and calculation of the exact Hellmann–Feynman forces after each MD step using the preconditioned conjugate techniques, and the Nosé dynamics for generating a canonical NVT ensemble [39]. Gaussian smearing (smearing width 0.1 eV) was used. The projector augmented wave (PAW) approach [40] within the generalized gradient approximation was utilized [41].
For first-principles electronic structure calculations, we utilized cutoff energies of 520.0 eV for the wave functions and 700.0 eV for the augmentation functions, as described in [16,30]. These values are notably higher than the corresponding default energies: ENMAX/Eaug = 400.0 eV/605.4 eV for O; ENMAX/Eaug = 240.3 eV/191.1 eV for Al; ENMAX/Eaug = 200.0 eV/454.7 eV for Mg; ENMAX/Eaug = 229.4 eV/355.8 eV for Sr; and ENMAX/Eaug = 187.2 eV/351.2 eV for Ba. The higher cutoff energies are necessary to describe the atoms well and to produce reliable results. Reasonably packed k-meshes were used for sampling the electronic wave functions, e. g. a 2 × 2 × 2 (8 k-points) in the Brillouin zone (BZ) of the sizable interface systems in the Monkhost-Pack approach [42]. For the simulations of the AE-segregated L-Al/γ-Al2O3{1 1 1} (denoted as L-Al/γ-Al2O3{1 1 1}AlAE) interfaces, a cutoff energy of 320.0 eV, and the Γ-point in the BZ were utilized. The latter was used due to the lack of periodicity of the whole system for one liquid/solid interface [16,30,39,43,44,45,46,47]. Tests using cutoff energies ranging from 200.0 eV to 400.0 eV proved availability of the settings.
We equilibrated those interface systems with full relaxation of the atomic coordinates for 4000 to 10,000 steps (1.5 femtosecond (fs) per step) at 1000 K, which is higher than the melting point of Al (933 K). The time-averaged technique was used for sampling the equilibrated interface systems over 3.0–4.5 picoseconds (ps) in order to secure statistically meaningful results [16,30,43,47].
3. Results
We first report our observation of the AIMD simulation processes. It was observed that the segregated AE atoms remained close to the γ-Al2O3{1 1 1} substrate surfaces and had no trend of moving into the liquid Al. This indicates stability of the AE-segregated interfaces, corresponding to the lower electronegativity of the AE atoms as compared to that of Al. Meanwhile, we also observed occasions in which Mg atoms moved into the oxide substrates (see Supplementary Materials Figure S1). These observations may relate to the formation of spinel from γ-alumina in Mg–Al liquid using liquid handling and casting [12,13]. We will not further discuss this issue in this paper.
At the first 0.5 ps, some AE atoms moved from one site to another at the surfaces of the substrates. Thereafter, they were in motion around the sites throughout the simulation, up to 15 ps. Analysis of the energies discloses that the systems reached thermal equilibrium within a short time (within 1.0 ps). Figure 1 shows snapshots of the equilibrated L-Al/γ-Al2O3{1 1 1}AlAE systems.
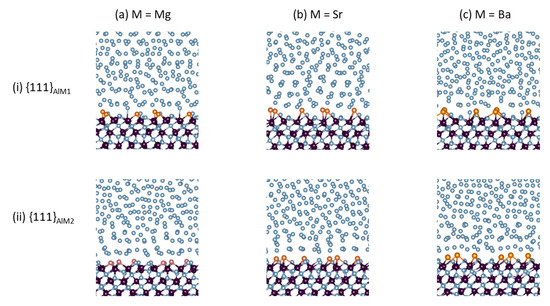
Figure 1.
Snapshots of the equilibrated L-Al/γ-Al2O3{1 1 1}AlAE interfaces. (a) L-Al/γ-Al2O3{1 1 1}AlMg, (b) L-Al/γ-Al2O3{1 1 1}AlSr, and (c) L-Al/γ-Al2O3{1 1 1}AlBa. Row (i) is for S1- and Row (ii) for S2-interfaces. The interatomic bonds between the metal atoms and the oxygen atoms in the substrates and at the substrate surfaces are displayed. The silvery spheres represent Al, dark brown spheres represent O, and the gold spheres represent the AE atoms.
At thermal equilibrium, the frames of the local structures of the L-Al/γ-Al2O3{1 1 1}AlAE interfaces (Figure 1) were similar to those of the authentic interfaces in [16]. The atoms in the γ-Al2O3 substrates are well ordered, whereas the Al atoms in middle of Al liquid parts display no apparent long-range ordering, instead behaving in a liquid-like way (Figure 1). Meanwhile, the segregated AE and the Al atoms adjacent to the substrates exhibited unusual behaviors. The AE atoms and the nearby Al atoms bonded to the O atoms at the alumina substrates. Moreover, the metal atoms near the interfaces displayed density variation in the direction perpendicular to the substrates, which is a phenomenon referred to as layering [16,39,44]. In order to assess the layering at the L-Al/γ-Al2O3{1 1 1}AlAE interfaces, we used atomic density profiles, which are defined in [18,19,20,44], for configurations summed over 3 ps of the L-Al/γ-Al2O3{1 1 1}AlAE interfaces. The obtained results are plotted in Figure 2.
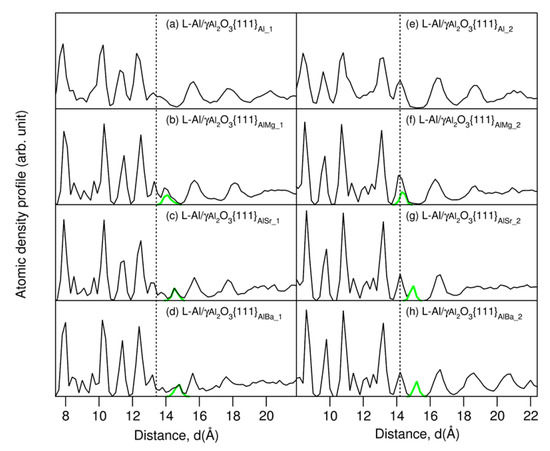
Figure 2.
Atomic density profiles of the AE-segregated L-Al/γ-Al2O3{1 1 1}AlAE interfaces and the parent L-Al/γ-Al2O3{1 1 1}Al (a,e) [16]. The black curves represent the L-Al/γ-Al2O3 interfaces for all atoms, and the green for the AE atoms. The dotted lines represent the terminating Al atoms.
The atomic density profiles verified our observations from the snapshots: there were ordered γ-Al2O3 substrates (sharp peaks), and liquid-like Al in the middle of the liquid parts (Figure 2). The liquid metal atoms near the substrates contained ordering along the direction perpendicular to the substrates (layering), and one parallel to the substrate (in-plane ordering). The segregated AE atoms were positioned close to the outermost oxygen atoms. Subtle differences at the two different interfaces, especially for the terminating AE and Al atoms, are apparent. At S1, the smaller Mg atoms are in the positions at the upper part of the termination layer (Figure 2b). The larger Sr and Ba atoms form an isolated atomic layer, which is positioned between the terminating Al layer and the first Al layer (Figure 2c,d). At S2, the smaller Mg atoms are also at the upper side of the single terminating Al peak. Meanwhile, the larger Sr and Ba atoms form isolated peaks between the terminating and first Al peaks (Figure 2g,h). Thus, the terminating metal layer at S2 (Figure 2e) becomes split, and the substrate surface becomes atomically rough as compared to that of the authentic interface (Figure 2e) [16].
Clearly, the distance between the AE atoms and the outmost O atoms is dependent on the atomic size of the segregated atoms (Figure 1 and Figure 2). The larger the atomic size is, the longer the distance from the outmost O layer to the positions of the AE ions, as shown in Figure 1 and Figure 2. According to the epitaxial nucleation model [17], the ordering of the terminating metal atoms at a liquid/solid interface is related to the intrinsic capability (potency) of the substrate to nucleate a solid in the liquid. Here, we analyze the behavior of the metal atoms neighboring the substrates. We integrated equilibrated configurations over 3 ps in order to obtain time-averaged atomic arrangements, which are shown in Figure 3. From the schematic atomic arrangements, the following information can be obtained:
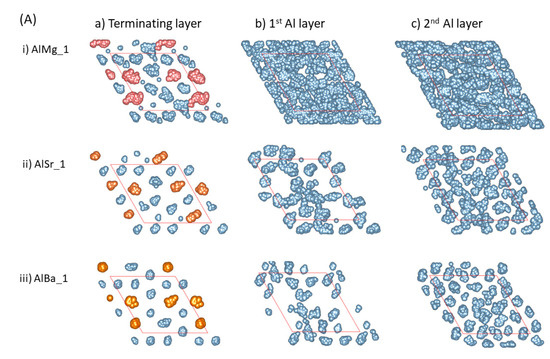
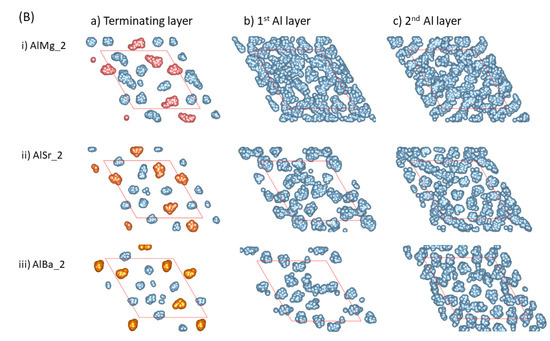
Figure 3.
The summed atomic configurations of the terminating Al/AE layer (a), the first Al layer (b) and the second Al layer (c) of the S1 ((A), top) and S2 (bottom, (B)) interfaces of L-Al/L-Al/γ-Al2O3 systems over 3 ps. The silvery spheres represent Al, the dark brown spheres represent O, and the gold spheres represent the AE atoms.
- (1)
- The AE and Al atoms at the terminating layer show strong localized characteristics, as compared with those metal atoms at the pristine L-Al/γ-Al2O3{1 1 1} interfaces [16].
- (2)
- The localization content of the terminating atoms increases from Mg to Sr to Ba. Figure 3i shows that only the atoms at the terminating Al/Mg layer exhibit some localized character, while the Al atoms at the first layer display delocalized, liquid-like behavior. There are vacancies on top of Sr and Ba atoms at the first Al layer, and localization exists at the second Al layers (Figure 3Aii, Aiii, Bii and Biii). This phenomenon may relate to the atomic sizes.
- (3)
- Prenucleation is weak even at the first metal layer for the Mg-segregated interfaces. For the Sr- and Ba-segregated interfaces, there are vacancies at the first Al layer and it thus, is rough. This causes amorphous arrangements of the Al atoms at the second layers. Analysis showed no long-range ordering at the third layer (not shown here).
Statistics of the interfacial O–M bonds between a substrate and a liquid or amorphous layer may give insight into the interfacial chemistry [16,30,33,47]. The interfacial bonding between terminating metal atoms and the outmost O atoms at the L-Al/γ- alumina interfaces with and without AE segregations was analyzed. There are four types of M coordination by O (Figure 4b,e). The obtained statistical results are plotted into Figure 4a.
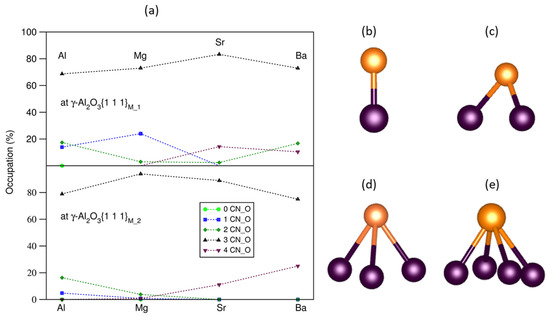
Figure 4.
The statistics of the occupation rates of the segregated AE at the O sites (O coordination numbers) (a), the schematic structures of AE-O (b), AE-2O (c), AE-3O (d), and AE-4O (e) bonding at the L-Al/γ-Al2O3 interfaces. In (b–e), the dark brown spheres represent O, and the gold spheres represent AE atoms.
The segregated AE atoms favor being on top of O triangles (over 70%) at both S1 and S2 interfaces (Figure 4d). At S1, about one quarter of the smaller Mg atoms are positioned on top of O (Figure 4b). There is a notable fraction of Ba atoms (about 20%) on top of four O atoms (Figure 4e). About 15% of Sr atoms are also positioned at the O bridges (Figure 4c). These relate to the size of the AE atoms.
Charge transfer between interfacial atoms can help to obtain a deeper insight into the interfacial interaction [16,30,47]. We investigated charges at the atomic sites at the AE-segregated L-Al/γ-alumina interfaces using the calculated electron density distributions via Bader’s model [48,49], which was implanted into this VASP code by Henkelman and colleagues [50]. The obtained results are plotted in Figure 5.
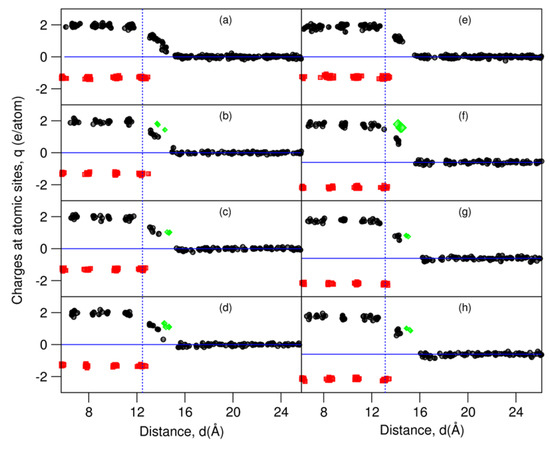
Figure 5.
Charges at the atomic sites at L-Al/γ-Al2O3 interfaces, by means of Bader analysis [44,45]. The black spheres represent charges at the Al sites, red at the O sites, and green at the AE sites. (a) L-Al/γ-Al2O3{1 1 1}Al-1, (b) L-Al/γ-Al2O3{1 1 1}AlMg-1, (c) L-Al/γ-Al2O3{1 1 1}AlSr-1, (d) L-Al/γ-Al2O3{1 1 1}AlBa-1, (e) L-Al/γ-Al2O3{1 1 1}Al-2, (f) L-Al/γ-Al2O3{1 1 1}AlMg-2, (g) L-Al/γ-Al2O3{1 1 1}AlSr-2, and (h) L-Al/γ-Al2O3{1 1 1}AlBa-2.
The O atoms in the substrates are negatively charged, with a value of −1.3 e/O, whereas the Al atoms in the substrates are positively charged (2.0 e/Al). This corresponds to the chemical formula (Al+2.02)2(O−1.34)3, as our calculations for the bulk oxides, which are shown in Table 1. The valences are smaller than those of the ionic model (Al+3)2(O−2)3, indicating some covalence in this oxide. The Al atoms away from the interfaces are electronically neutral.
As shown in Figure 5, the terminating metal (Al and AE) atoms are partially charged. The charges of the Al atoms decrease with the distance from the outmost oxygen atoms/ions, in agreement with the bond theory [51,52]. The charges at the AE sites are higher than those at the Al sites at the same z-positions.

Table 1.
Electronic configurations and electronegativity, χ, of the alkaline earth elements Mg, Sr and Ba, as well as Al. These elements have s, p valence electrons. The (averaged) bond lengths between the metals and oxygen in their oxides, MgO, SrO, and BaO of the NaCl-type structure, as well as α-Al2O3, with a trigonal lattice, as discussed in the literature [53,54], are included. Charges at the atomic sites from present calculations are shown in the formula.
Table 1.
Electronic configurations and electronegativity, χ, of the alkaline earth elements Mg, Sr and Ba, as well as Al. These elements have s, p valence electrons. The (averaged) bond lengths between the metals and oxygen in their oxides, MgO, SrO, and BaO of the NaCl-type structure, as well as α-Al2O3, with a trigonal lattice, as discussed in the literature [53,54], are included. Charges at the atomic sites from present calculations are shown in the formula.
| Element | Elec. Config. | Χ | Formula of Oxides | d(M–O) (Å) |
|---|---|---|---|---|
| Al | [Ne] 3s2 3p1 | 1.61 | Al+2.022O−1.343 | 1.92 [53] |
| Mg | [Ne] 3s2 3p0 | 1.31 | Mg+1.66O−1.66 | 2.11 [53] |
| Sr | [Kr] 5s2 5p0 | 0.95 | Sr +1.35O−1.35 | 2.57 [54] |
| Ba | [Xe] 6s2 6p0 | 0.89 | Ba+1.36O−1.36 | 2.76 [54] |
4. Discussion
The present simulations revealed that the segregated AE atoms are chemically bonded to the outmost oxygen atoms at the L-Al/γ-Al2O3{1 1 1} interfaces, at a temperature above the melting point of Al. Such segregation of the AE atoms at the interfaces can be regarded as reduction–oxidation (redox) reactions. As shown in Table 1, the valence bands of Al and the AE atoms are composed of s, p electrons. The electronegativity of Al is notably higher than that of each of the AE elements, which makes the redox actions occur. Moreover, Al has three valence electrons, whereas the AE atoms each have two (Table 1).
Table 1 also lists the (averaged) metal–oxygen (M–O) bond lengths in the bulk oxides [53,54]. Clearly, the M–O bond increases from Al to Mg, to Sr, and to Ba. Based on the M–O bond lengths, we can obtain the ionic sizes of the metals R(Mm+) = d(M–O) − R(O−q), where R(O−q) = 1.4 Å, as reported in the literature [55,56]. We thus build relationships between the splitting of the terminating metal layer and the AE ionic sizes at the L-Al/γ-Al2O3{1 1 1}AlAE interfaces. The obtained results are plotted in Figure 6.
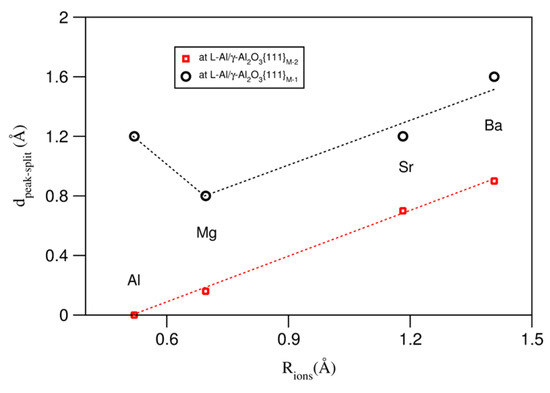
Figure 6.
Relationships between distances of the splitting of the termination metal layers at L-Al/γ-Al2O3 interfaces and the ionic sizes of the AE atoms.
There are almost-linear relationships between the ionic size and the splitting of the terminating metal layers at both L-Al/γ-Al2O3{1 1 1} interfaces, except for Mg at S1, where the smaller Mg atoms/ions are positioned at the top of the terminating layer, which, in fact, reduces the peak splitting. This is partially due to the valence difference between Mg and Al.
Segregation of the AE atoms at the L-Al/γ-Al2O3{1 1 1}AlAE2 interface roughens the flat termination metal layer (Figure 2e). Correspondingly, prenucleation at the segregated substrate surfaces turns out to be hindered. This results in the potencies of the two authentic different substrate surfaces becoming minimized to the same level. Naturally, such segregated substrate surfaces of minimized potency require a large drive force to nucleate the solid phase. Meanwhile, the number of solidification sites is significantly enhanced. This indicates a possibility of achieving cast parts/alloys of finer microstructures if there is no significant concentration of more potent particles in the liquid, according to the EPI theory [4,5].
The present simulations disclosed a possibility of segregations of more chemically active (less electronegative) impurities at a liquid/oxide interface via redox reactions. Moreover, the simulations also showed that impure atoms of larger atomic sizes than that of the liquid metal may cause higher occurrences of atomic roughness of the substrate surface. This may help us design grain refiners for the solidification of various liquid metals/alloys. Moreover, this influence of segregated impurities on the atomic ordering at liquid-metal/solid interfaces may play a role in thermodynamics investigations on the growth of solid particles in a liquid during solidification [57,58]. This knowledge regarding the influences of segregated impurities at a solid/liquid metal interface might be useful for obtaining insight into the role of impurities segregated at metal–ceramics interfaces [59], the wetting phenomenon, and, furthermore, into metal/ceramic welding, surface treatments, etc. [60]. Broadly, the knowledge obtained here may further help us control solidification processes, lower manufacturing costs, and benefit our circular economy.
5. Summary
We performed ab initio molecular dynamics simulations for the AE-segregated L-Al/γ-Al2O3{1 1 1} interfaces. The AIMD simulations provided us the following conclusions:
- (1)
- The AE-atom-segregated L-Al/γ-Al2O3{1 1 1} interfaces exhibit localized characteristics at the interfaces.
- (2)
- Chemically, the AE atoms are positively charged and bonded to the outmost O atoms.
- (3)
- The segregated AE atoms are positioned dominantly on O triangles of the γ-Al2O3{1 1 1} substrates, a condition which reconstructs atoms adjacent to the substrates and causes atomic roughness of the substrate surfaces.
- (4)
- The sizes of the AE ions significantly affect the atomic ordering at the interfaces.
- (5)
- Prenucleation at the two different authentic substrates causes them to become weakened and minimized to the same level.
- (6)
- The obtained information herein enriches our understanding of the effects of impurity segregation at liquid metal/solid interfaces, and further helps to manipulate heterogeneous nucleation via controlling impurity segregation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/met13040761/s1, Figure S1: Schematic presentation of one segregated Mg atom (green sphere) moving into the oxide substrate during the ab initio molecular dynamics simulations. The large dark brown spheres represent O, middle silver spheres represent Al, and orange spheres represent Mg.
Author Contributions
C.F. conducted ab initio simulations, data analysis, visualization, and original draft writing; Z.F. conducted initiation of the research, funding acquisition, and supervision. Both authors contributed to the review and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been funded by the EPSRC of the UKRI under the grant numbers EP/N007638/1 and EP/S005102/1, respectively.
Data Availability Statement
Supplementary Materials are provided.
Acknowledgments
The authors thank B. Cantor, as well as I. Chang, Y. Wang, and S.H. Wang (BCAST) for beneficent discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Turnbull, D. Kinetics of heterogeneous nucleation. J. Chem. Phys. 1950, 18, 198–203. [Google Scholar] [CrossRef]
- Murty, B.S.; Kori, S.A.; Chakraborty, M. Grain refinement of aluminium and its alloys by heterogeneous nucleation and alloying. Intern. Mater. Rev. 2002, 47, 3–29. [Google Scholar] [CrossRef]
- Kelton, K.F.; Greer, A.L. Nucleation in Condensed Matter: Applications in Materials and Biology; Elsevier Science: Oxford, UK, 2010. [Google Scholar]
- Fan, Z.; Mendis, C. Heterogeneous nucleation, grain initiation and grain refinement of Mg-alloys. In Proceedings of the 11th International Conference on Magnesium Alloys and Their Applications, Beaumont Estate, Old Windsor, UK, 24–27 July 2018. [Google Scholar]
- Fan, Z.; Gao, F.; Jiang, B.; Que, Z.P. Impeding nucleation for more significant grain refinement. Sci. Rep. 2020, 10, 9448. [Google Scholar] [CrossRef] [PubMed]
- Cibula, A. The grain refinement of aluminium alloy castings by addition of titanium and boron. J. Inst. Met. 1951, 80, 1–16. [Google Scholar]
- Greer, A.L.; Bunn, A.M.; Tronche, A.; Evans, P.V.; Bristow, D.J. Modelling of inoculation of metallic melts: Application to grain refinement of aluminium by Al-Ti-B. Acta Mater. 2000, 48, 2823–2835. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Zhang, Y.; Qin, T.; Zhou, X.R.; Thompson, G.E.; Pennycook, T.; Hashimoto, T. Grain refining mechanism in the Al/Al–Ti–B system. Acta Mater. 2015, 84, 292–304. [Google Scholar] [CrossRef]
- Greer, A.L. Overview: Application of heterogeneous nucleation in grain-refining of metals. J. Chem. Phys. 2016, 145, 211704. [Google Scholar] [CrossRef]
- Impey, S.A.; Stephenson, D.J.; Nicholls, J.R. Mechanism of scale growth on liquid aluminium. J. Mater. Sci. Technol. 1988, 4, 1126–1132. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.-T.; Fan, Z. Oxidation of aluminium alloy melts and inoculation by oxide particles. Trans. Indian Inst. Met. 2012, 65, 653–661. [Google Scholar] [CrossRef]
- Kim, K. Formation of endogenous MgO and MgAl2O4 particles and their possibility of acting as substrate for heterogeneous nucleation of aluminum grains. Surf. Interface Anal. 2015, 47, 429–438. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Xia, M.; Arumuganathar, S. Enhanced heterogeneous nucleation in AZ91D alloy by intensive melt shearing. Acta Mater. 2009, 57, 4891–4901. [Google Scholar] [CrossRef]
- Verwey, E.J.W. The crystal structure of γ-Fe2O3 and γ-Al2O3. Z. Kristallogr. 1935, 91, 65–69. [Google Scholar] [CrossRef]
- Smrčok, L.; Langer, L.; Krestan, J. γ-Alumina: A single crystal X-ran diffraction study. Acta Cryst. C 2006, 62, i83–i84. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.M.; Yasmin, S.; Fan, Z. Interfacial interaction and prenucleation at liquid-Al/γ-Al2O3{1 1 1} interfaces from ab initio molecular dynamics simulations. J. Phys. Commun. 2021, 5, 015007. [Google Scholar] [CrossRef]
- Fan, Z. An epitaxial model for heterogeneous nucleation on potent substrates. Metall. Mater. Trans. A 2013, 44, 1409–1418. [Google Scholar] [CrossRef]
- Men, H.; Fan, Z. Prenucleation induced by crystalline substrates. Metall. Mater. Trans. A 2018, 49, 2766–2777. [Google Scholar] [CrossRef]
- Men, H.; Fan, Z. Prenucleation at the liquid/substrate interfaces: An overview. Metals 2022, 12, 1704. [Google Scholar] [CrossRef]
- Fang, C.M.; Men, H.; Fan, Z. Effect of substrate chemistry on prenucleation. Metall. Mater. Trans. A 2018, 49, 6231–6242. [Google Scholar] [CrossRef]
- Bunn, A.M.; Schumacher, P.; Kearns, M.A.; Boothroy, C.B.; Greer, A.L. Grain refinement by Al-Ti-B alloys in aluminium melts: A study of the mechanisms of poisoning by zirconium. Mater. Sci. Technol. 1999, 15, 1115–1123. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, C.M.; Zhou, L.; Hashimoto, T.; Zhou, X.; Ramasse, Q.M.; Fan, Z. Mechanism for Zr poisoning of Al-Ti-B based grain refiners. Acta Mater. 2019, 164, 428–439. [Google Scholar] [CrossRef]
- Wang, Y.; Que, Z.P.; Hashimoto, T.; Zhou, X.R.; Fan, Z. Mechanism for Si poisoning of Al-Ti-B grain refiners in Al alloys. Metal. Mater. Trans. A 2020, 51, 5743–5757. [Google Scholar] [CrossRef]
- Wang, S.H.; Wang, Y.; Ramasse, Q.; Fan, Z. The nature of native MgO in Mg and its alloys. Metal. Mater. Trans. A 2020, 51, 2957–2974. [Google Scholar] [CrossRef]
- Ma, S.D.; Dong, Z.D.; Zong, Z.F.; Jing, T.; Dong, H.B. Solute-adsorption enhanced heterogeneous nucleation: The effect of Cu adsorption on α-Al at the sapphire substrate. Phys. Chem. Chem. Phys. 2021, 23, 5270–5282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.H.; Fang, C.M.; Que, Z.P.; Hashimoto, T.; Zhou, X.R.; Ramasse, Q.M.; Fan, Z. Manipulating nucleation potency of substrates by interfacial segregation. Metals 2022, 12, 1636. [Google Scholar] [CrossRef]
- Ma, S.D.; Yan, R.; Zong, N.F.; Davidchack, R.L.; Jing, T.; Dong, H.B. Unveiling the influence of interfacial bonding and dynamics on solid/liquid interfacial structures: An ab initio molecular dynamics study of (0001) sapphire-liquid Al interfaces. Phys. Rev. Mater. 2020, 4, 023401. [Google Scholar] [CrossRef]
- Zhang, G.C.; Hu, T.; Shuai, S.S.; Chen, C.Y.; Xu, S.Z.; Yu, J.B.; Ren, W.; Wang, J.; Ren, Z.M. Ab-initio molecular dynamics study of heterogeneous nucleation at the liquid-Y/α-Al2O3 interface. Comput. Mater. Sci. 2023, 217, 111899. [Google Scholar] [CrossRef]
- Shen, G.; Lyu, X.M.; Zhao, S.Y.; You, C.; Wang, X.W.; Chen, M.F. The first-principles research of Cao and MgO particulate heterogeneous nucleation in Mg alloys. Appl. Surf. Sci. 2022, 593, 153224. [Google Scholar] [CrossRef]
- Fang, C.M.; Fan, Z. Effects of Segregation of Sc, Y and La Atoms on Prenucleation at the Interfaces between Liquid-Al and γ-Al2O3{1 1 1}. Metals 2022, 12, 1550. [Google Scholar] [CrossRef]
- Kang, J.; Zhu, J.; Curtis, C.; Blake, D.; Glatzmaier, G.; Kim, Y.-H.; Wei, S.-H. Atomically abrupt liquid-oxide interface stabilized by self-regulated interfacial defects: The case of Al/Al2O3 interfaces. Phys. Rev. Lett. 2012, 108, 226105. [Google Scholar] [CrossRef]
- Yan, R.; Sun, W.Z.; Ma, S.D.; Jing, T.H.B.; Dong, H.B. The orientation dependence of liquid ordering at α-Al2O3/Al solid-liquid interfaces: A molecular dynamics study. Comput. Mater. Sci. 2020, 174, 109489. [Google Scholar] [CrossRef]
- Fang, C.M.; Fan, Z. Prenucleation at the liquid-Al/α-Al2O3 and the liquid-Al/MgO interfaces. Comput. Mater. Sci. 2020, 171, 109258. [Google Scholar] [CrossRef]
- Oh, S.H.; Kauffmann, Y.; Scheu, C.; Kaplan, W.D.; Rühle, N. Ordered liquid aluminium at the sapphire. Science 2005, 310, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, Y.; Oh, S.H.; Koch, C.T.; Hashibon, A.; Scheu, C.; Rühle, M.; Kaplan, W.D. Quantitative analysis of layering and in-plane ordering structural ordering at an alumina-aluminum solid liquid interface. Acta Mater. 2011, 59, 4378–4386. [Google Scholar] [CrossRef]
- Curiotto, S.; Chien, H.; Meltzman, H.; Wynblatt, P.; Rohrer, G.S.; Kaplan, W.D.; Chatain, D. Orientation relationships of copper crystals on cplane sapphire. Acta Mater. 2011, 59, 5320–5331. [Google Scholar] [CrossRef]
- Fang, C.M.; Fan, Z. Prenucleation at the interface between MgO and liquid magnesium: An ab initio molecular dynamics study. Metal. Mater. Trans. A 2020, 51, 788–797. [Google Scholar] [CrossRef]
- Fang, C.M.; Fan, Z. Atomic ordering at the Liquid-Al/MgAl2O4{1 1 1} Interfaces: Ab initio molecular dynamics simulations. Metal. Mater. Trans. A 2020, 51, 6318–6326. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17978. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Brostow, W.; Hagg-Lodbland, H.E. Materials: Introduction and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Hashibon, A.; Adler, J.; Finnis, M.W.; Kaplan, W.D. Atomistic study of structural correlations at a liquid-solid interface. Comput. Mater. Sci. 2002, 24, 443–452. [Google Scholar] [CrossRef]
- Wearing, D.; Horsfield, A.P.; Xu, W.W.; Lee, P.D. Which wets TiB2 inoculant particles: Al or Al3Ti? J. Alloys Compd. 2016, 664, 460–468. [Google Scholar] [CrossRef]
- Wang, J.S.; Horsfield, A.P.; Schwingenschlögl, U.; Lee, P.D. Heterogeneous nucleation of solid Al from the melt by TiB2 and Al3Ti: An ab initio molecular dynamics study. Phys. Rev. B 2010, 82, 184203. [Google Scholar] [CrossRef]
- Fang, P.X.W.; Nihtianov, S.; Sberma, P.; Fang, C.M. Interfaces between crystalline Si and amorphous B: Interfacial interactions and change barriers. Phys. Rev. B 2021, 103, 075301. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum-theory of molecular-structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. A bonded path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 254–360. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Brown, I.D. Bond valence-a simple structural model for inorganic chemistry. Chem. Soc. Rev. 1978, 7, 359–376. [Google Scholar] [CrossRef]
- Fiquet, G.; Richet, P.; Montagnac, G. High-temperature thermal expansion of lime, periclase, corundum and spinel. Phys. Chem. Min. 1999, 17, 103–111. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. The Structure of Crystals, 2nd ed.; Reinhold Publishing Corporation: New York, NY, USA, 1935. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Cryst. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Bollada, P.C.; Men, M.; Fang, C.M.; Jimack, P.K.; Fan, Z.; Mullis, A.M. A novel route to the coupling of molecular dynamics and phase-field simulations of crystal growth. IOP Conf. Series 2019, 529, 012032. [Google Scholar] [CrossRef]
- Bollada, P.C.; Jimack, P.K.; Mullis, A.M. Towards a physically consistent phase-field model for alloy solidification. Metal 2022, 12, 272. [Google Scholar] [CrossRef]
- Singh, G.; Yu, Y.; Ernst, F.; Raj, R. Shear strength and sliding at a metal-ceramic (aluminium-spinel) interface at ambient and elevated temperatures. Acta Mater. 2007, 55, 3049–3057. [Google Scholar] [CrossRef]
- Karakozov, E.S.; Konyshov, G.; Musin, R.S. Fundamentals of welding metals to ceramic materials. J. Welding Intern. 1993, 7, 991–996. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).