
First-Principles Computation of Microscopic Mechanical Properties and Atomic Migration Behavior for Al4Si Aluminum Alloy

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
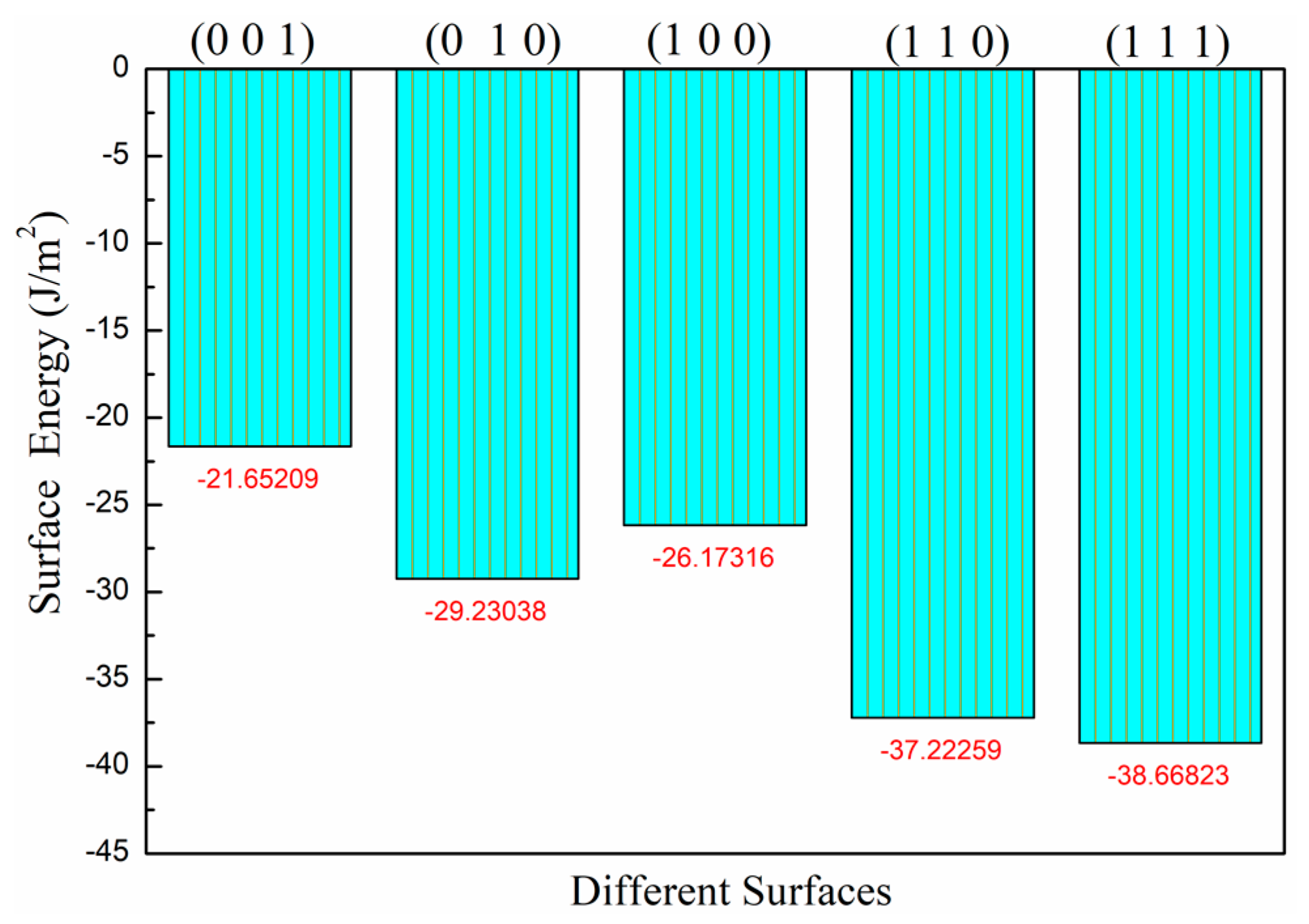
3.1. Surface Stability
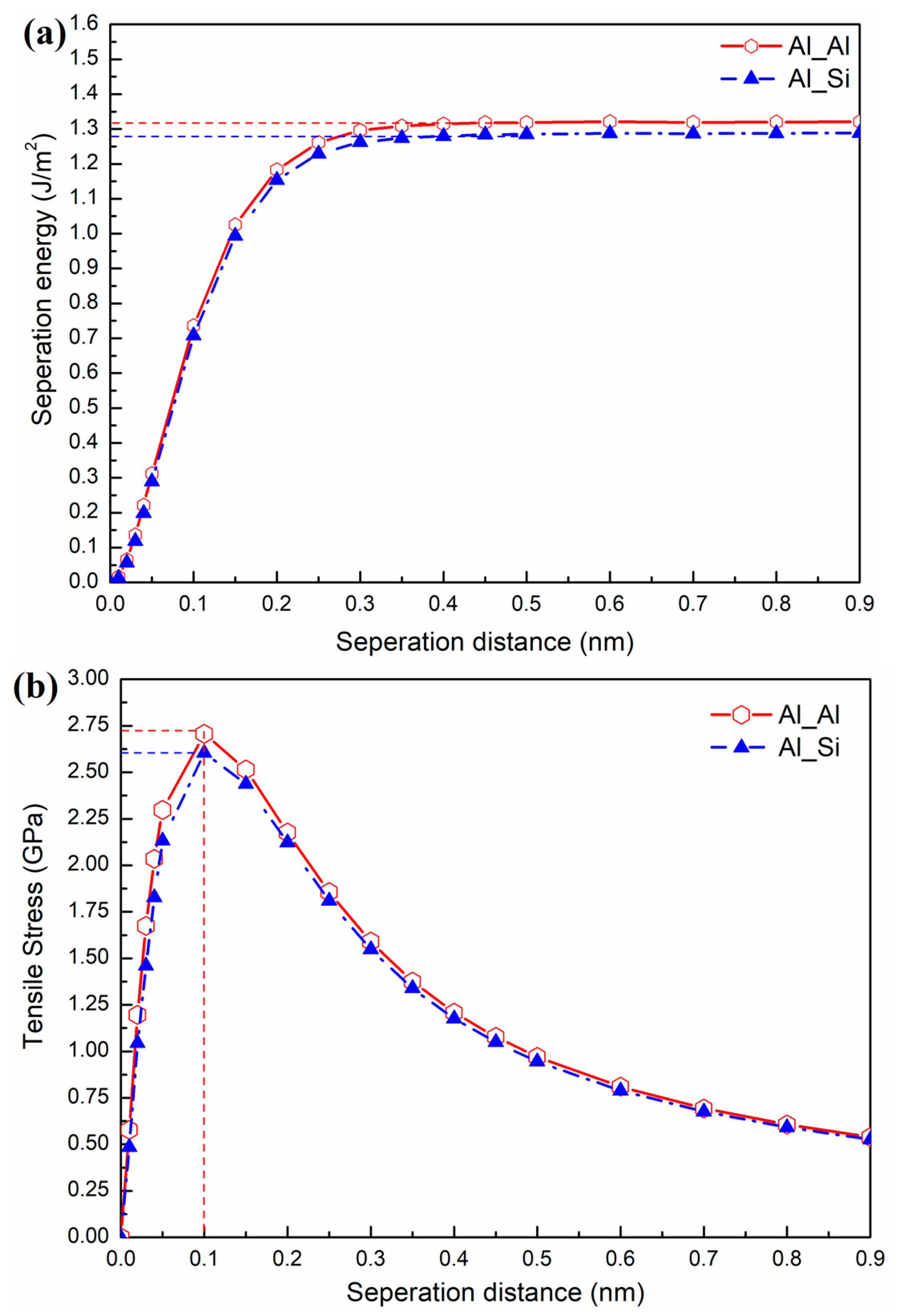
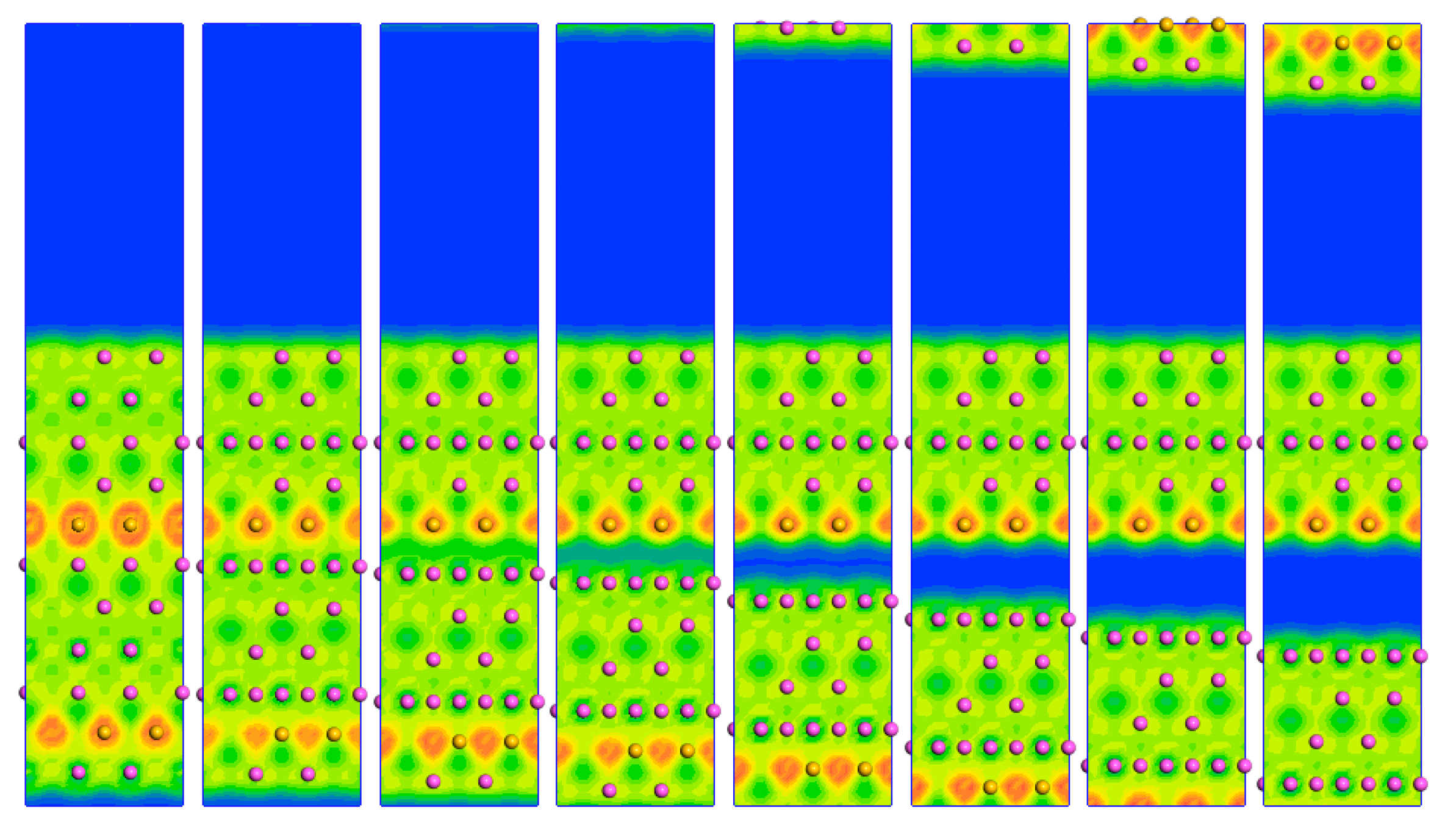
3.2. Micro-Mechanical Property
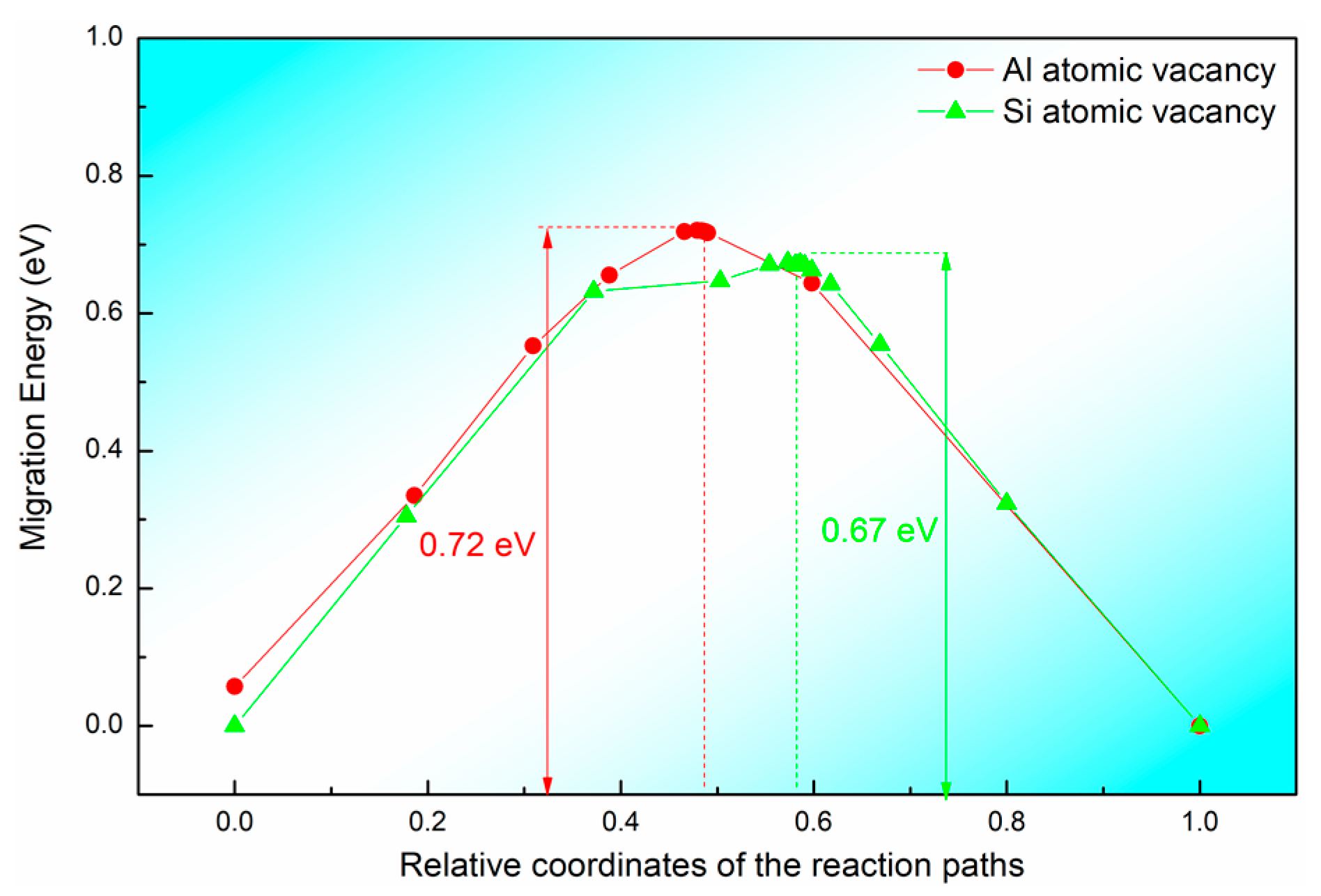
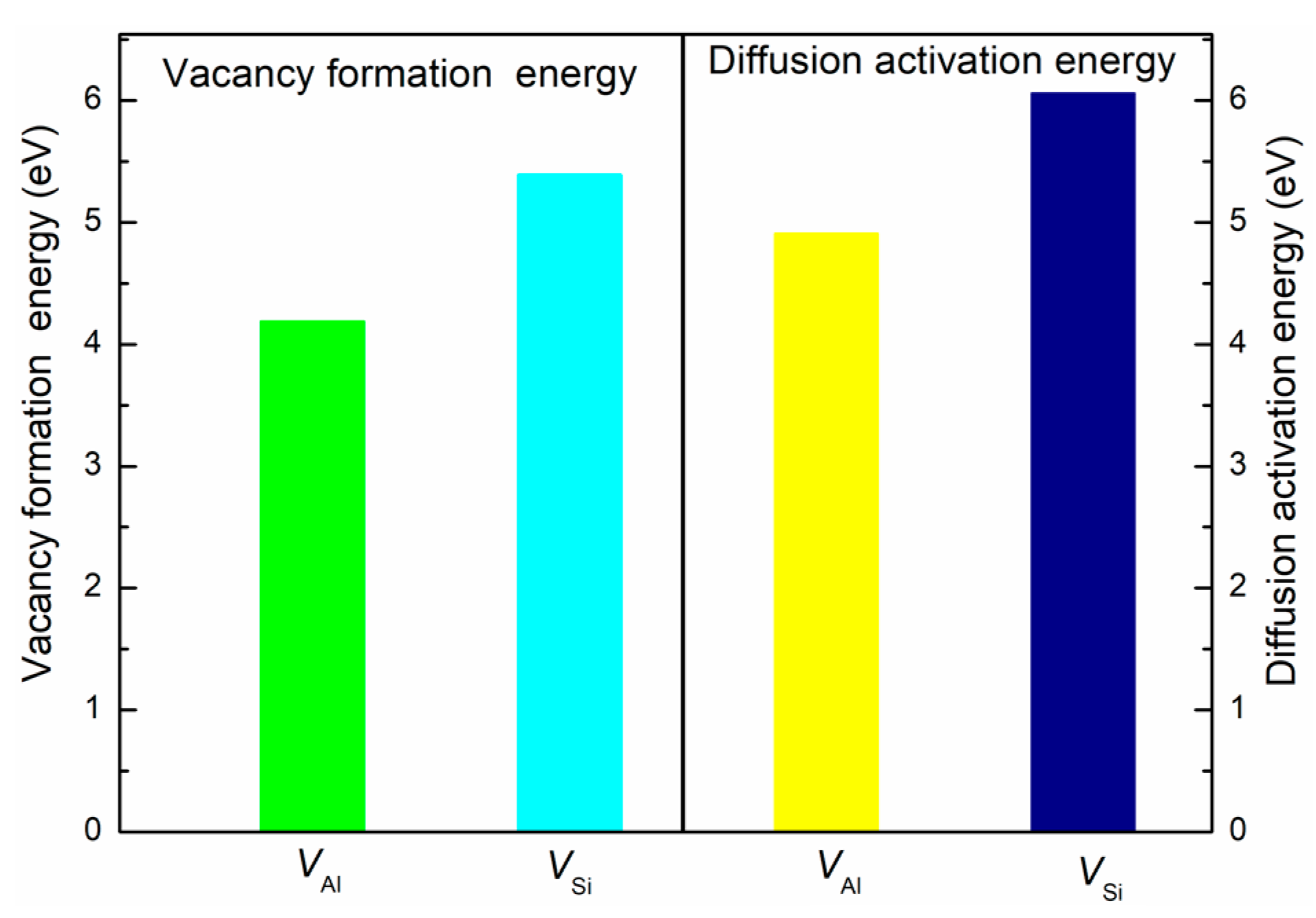
3.3. Diffusion Behavior
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Marquis, E.A.; Seidman, D.N.; Asta, M.; Woodward, C. Composition Evolution of Nanoscale Al3Sc Precipitates in an Al-Mg-Mn-Sc Alloy: Experiments and Computations. Acta Mater. 2006, 54, 119–130. [Google Scholar] [CrossRef]
- Mortsell, E.A.; Marioara, C.D.; Andersen, S.J.; Ringdalen, I.G.; Friis, J.; Wenner, S.; Royset, J.; Reiso, O.; Holmestad, R. The effects and behaviour of Li and Cu alloying agents in lean Al-Mg-Si alloys. J. Alloys Compd. 2017, 699, 235–242. [Google Scholar] [CrossRef]
- Biswas, A.; Siegel, D.J.; Wolverton, C.; Seidman, D.N. Precipitates in Al-Cu alloys revisited: Atom-probe tomographic experiments and first-principles calculations of compositional evolution and interfacial segregation. Acta Mater. 2011, 59, 6187–6204. [Google Scholar] [CrossRef]
- Topcu, I.; Gulsoy, H.O.; Kadioglu, N.; Gulluoglu, A.N. Processing and mechanical properties of B4C reinforced Al matrix composites. J. Alloys Compd. 2009, 482, 516–521. [Google Scholar] [CrossRef]
- Williams, J.C.; Starke, E.A., Jr. Progress in structural materials for aerospace systems. Acta Mater. 2003, 51, 5775–5799. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, X.; Li, H.; Zhao, Y.; Huang, Y.; Liu, Y.; Xiao, Z. Interfacial properties and fracture behavior of the L12-Al3Sc||Al interface: Insights from a first-principles study. Appl. Surf. Sci. 2020, 515, 146017. [Google Scholar] [CrossRef]
- Huang, J.; Li, M.; Liu, Y.; Chen, J.; Lai, Z.; Hu, J.; Zhou, F.; Zhu, J. A first-principles study on the doping stability and micromechanical properties of alloying atoms in aluminum matrix. Vacuum 2023, 207, 111596. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Y.; Liu, Y.; Xiao, Z. A comprehensive study of the L12-Al3Nb/Al interface properties using first-principles calculations. J. Mater. Res. Technol. 2020, 9, 12428–12442. [Google Scholar] [CrossRef]
- Getmanskii, I.V.; Koval, V.V.; Boldyrev, A.I.; Minyaev, R.M.; Minkin, V.I. Computationally Designed Crystal Structures ofhe Supertetrahedral Al4X (X = B, C, Al, Si) Solids. J. Phys. Chem. A 2019, 123, 267–271. [Google Scholar] [CrossRef]
- Pei, X.; Yuan, M.N.; Han, F.Z.; Wei, Z.Y.; Ma, J.; Wang, H.L.; Shen, X.Q.; Zhou, X.S. Investigation on tensile properties and failure mechanism of Al(111)/Al3Ti(112) interface using the first-principles method. Vacuum 2022, 196, 110784. [Google Scholar] [CrossRef]
- Peng, M.J.; Wang, R.F.; Wu, Y.J.; Yang, A.C.; Duan, Y.H. Elastic anisotropies, thermal conductivities and tensile properties of MAX phases Zr2AlC and Zr2AlN: A first-principles calculation. Vacuum 2022, 196, 110715. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.Z.; Zhang, S.Y.; Song, X.Q.; Wang, Y.X.; Chen, Z. First principles study of stability, electronic structure and fracture toughness of Ti3SiC2/TiC interface. Vacuum 2022, 196, 110745. [Google Scholar] [CrossRef]
- Wang, D.; Xiao, Z. Revealing the Al/L12-Al3Zr inter-facial properties: Insights from first-principles calculations. Vacuum 2022, 195, 110620. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Xu, L.L.; Zheng, H.F.; Xu, B.; Liu, G.Y.; Zhang, S.L.; Zheng, H.B. Suppressing Nonradiative Recombination by Electron-Donating Substituents in 2D Conjugated Triphenylamine Polymers toward Efficient Perovskite Optoelectronics. Nano Lett. 2023, 23, 1954–1960. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1998, 77, 3865–3868. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Marsman, M.; Paier, J.; Stroppa, A.; Kresse, G. Hybrid functionals applied to extended systems. J. Phys. Condens. Matter 2008, 20, 064201. [Google Scholar] [CrossRef]
- Deane, K.; Kampe, S.L.; Swenson, D.; Sanders, P.G. Precipitate Evolution and Strengthening in Supersaturated Rapidly Solidified Al-Sc-Zr Alloys. Met. Mater. Trans. A 2017, 48, 2030–2039. [Google Scholar] [CrossRef]
- Mantina, M.; Wang, Y.; Arroyave, R.; Liu, L.Q.C.Z.K.; Wolverton, C. First-Principles Calculation of Self-Diffusion Coefficients. Phys. Rev. Lett. 2008, 100, 215901. [Google Scholar] [CrossRef]
- Rose, J.H.; Ferrante, J.; Smith, J.R. Universal bnding energy curves for metals and bimetallic interfaces. Phys. Rev. Lett. 1981, 47, 675–678. [Google Scholar] [CrossRef]
- Yuryev, P.O.; Baranov, V.N.; Orelkina, T.A.; Bezrukikh, A.I.; Voroshilov, D.S.; Murashkin, M.Y.; Partyko, E.G.; Konstantinov, I.L.; Yanov, V.V.; Stepanenko, N.A. Investigation the structure in cast and deformed states of aluminum alloy, economically alloyed with scandium and zirconium. Int. J. Adv. Manuf. Technol. 2021, 115, 263–274. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, P.; Shao, D.; Wang, R.; Cao, L.; Zhang, J.; Liu, G.; Chen, B.; Sun, J. The influence of Sc solute partitioning on the microalloying effect and mechanical properties of Al-Cu alloys with minor Sc addition. Acta Mater. 2016, 119, 68–79. [Google Scholar] [CrossRef]
- Liang, S.S.; Wen, S.P.; Wu, X.L.; Huang, H.; Gao, K.Y.; Nie, Z.R. The synergetic effect of Si and Sc on the thermal stability of the precipitates in AlCuMg alloy. Mater. Sci. Eng. A 2020, 78, 139319. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Huang, J.T.; Li, M.W.; Chen, J.Y.; Cheng, Y.; Lai, Z.H.; Hu, J.; Zhou, F.; Qu, N.; Liu, Y.; Zhu, J.C. Electronic structure and atomic migration of the fourth, fifth, and sixth period atoms in aluminium alloys: First principles calculation. Vacuum 2023, 210, 111823. [Google Scholar] [CrossRef]
- Mantina, M.; Shang, S.L.; Wang, Y.; Chen, L.Q.; Liu, Z.K. 3D transition metal impurities in aluminum: A first-principles study. Phys. Rev. B 2009, 80, 184111. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, H.; Xiao, S.; Tang, J.; Deng, L.; Hu, W. First-principles calculation of self-diffusion coefficients in Ni3Al. J. Alloys Compd. 2014, 612, 361–364. [Google Scholar] [CrossRef]
- Zhou, B.-C.; Shang, S.-L.; Wang, Y.; Liu, Z.-K. Diffusion coefficients of alloying elements in dilute Mg alloys: A comprehensive first-principles study. Acta Mater. 2016, 103, 573–586. [Google Scholar] [CrossRef]
- Huber, L.; Elfimov, I.; Rottler, J.; Militzer, M. Ab initio calculations of rare-earth diffusion in magnesium. Phys. Rev. B 2012, 85, 4214–4218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | EForm (eV) | EJ (eV) | EQ (eV) |
|---|---|---|---|
| VAl | 4.18 | 0.72 | 4.90 |
| VSi | 5.39 | 0.67 | 6.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Xue, J.; Li, M.; Cheng, Y.; Lai, Z.; Hu, J.; Zhou, F.; Qu, N.; Liu, Y.; Zhu, J. First-Principles Computation of Microscopic Mechanical Properties and Atomic Migration Behavior for Al4Si Aluminum Alloy. Metals 2023, 13, 1622. https://doi.org/10.3390/met13091622
Huang J, Xue J, Li M, Cheng Y, Lai Z, Hu J, Zhou F, Qu N, Liu Y, Zhu J. First-Principles Computation of Microscopic Mechanical Properties and Atomic Migration Behavior for Al4Si Aluminum Alloy. Metals. 2023; 13(9):1622. https://doi.org/10.3390/met13091622
Chicago/Turabian StyleHuang, Jingtao, Jingteng Xue, Mingwei Li, Yuan Cheng, Zhonghong Lai, Jin Hu, Fei Zhou, Nan Qu, Yong Liu, and Jingchuan Zhu. 2023. "First-Principles Computation of Microscopic Mechanical Properties and Atomic Migration Behavior for Al4Si Aluminum Alloy" Metals 13, no. 9: 1622. https://doi.org/10.3390/met13091622
APA StyleHuang, J., Xue, J., Li, M., Cheng, Y., Lai, Z., Hu, J., Zhou, F., Qu, N., Liu, Y., & Zhu, J. (2023). First-Principles Computation of Microscopic Mechanical Properties and Atomic Migration Behavior for Al4Si Aluminum Alloy. Metals, 13(9), 1622. https://doi.org/10.3390/met13091622